UNIVERSIDADE DE LISBOA
FACULDADE DE FARMÁCIA
DEPARTAMENTO DE BIOQUÍMICA E BIOLOGIA HUMANA
Disturbed Protein Arginine
Methylation in Hyperhomocysteinemia
Ruben Miguel Costa Fernandes Esse
DOUTORAMENTO EM FARMÁCIA BIOQUÍMICA
UNIVERSIDADE DE LISBOA
FACULDADE DE FARMÁCIA
DEPARTAMENTO DE BIOQUÍMICA E BIOLOGIA HUMANA
Disturbed Protein Arginine
Methylation in Hyperhomocysteinemia
Ruben Miguel Costa Fernandes Esse
DOUTORAMENTO EM FARMÁCIA BIOQUÍMICA
UNIVERSIDADE DE LISBOA
FACULDADE DE FARMÁCIA
DEPARTAMENTO DE BIOQUÍMICA E BIOLOGIA HUMANA
Disturbed Protein Arginine
Methylation in Hyperhomocysteinemia
Distúrbio da Metilação Proteica em Resíduos de
Arginina na Hiperhomocisteinémia
Ruben Miguel Costa Fernandes Esse
Dissertação apresentada à Faculdade de Farmácia da Universidade de Lisboa para obtenção do grau de Doutor em Farmácia (especialidade: Bioquímica)
Supervisor: Prof. Doctor Rita Azevedo e Castro
Co-supervisors: Prof. Doctor Maria Isabel Tavares de Almeida Prof. Doctor Henk J. Blom
Lisboa 2013
The studies presented in this thesis were performed at the Metabolism and Genetics Group, iMed. UL (Research Institute for Medicines and Pharmaceutical Sciences), and at the Department of Biochemistry and Human Biology, Faculdade de Farmácia da Universidade de Lisboa, Portugal, under the supervision of Prof. Doctor Rita Azevedo e Castro and Prof. Doctor Maria Isabel Tavares de Almeida, and at the Department of Clinical Chemistry, Metabolic Unit, Free University Medical Centre, Amsterdam, The Netherlands, under the supervision of Prof. Doctor Henk J. Blom.
This work was finantially supported by Fundação para a Ciência e a Tecnologia (F.C.T.), Lisboa, Portugal (SFRH/BD/48585/2008).
De acordo com o disposto no ponto 1 do artigo n.º 41 do Regulamento de Estudos Pós-Graduados da Universidade de Lisboa, deliberação n.º 93/2006, publicada em Diário da República – II série n.º 153 – de 5 de Julho de 2003, o autor desta dissertação declara que participou na conceção e execução do trabalho experimental, interpretação dos resultados obtidos e redação dos manuscritos.
Aos meus pais e irmã...
Interviewer: It’s all like The Doors’ song “Break on Through to the Other Side”. Patti Smith: Yeah, and then after you break on through to the other side, then you break on through to the
other side, and the other side, and the other side… I mean, our point is that you spend your whole life keep breaking on through. You can’t just break on through once and think ‘Well I’ve made it, I’ve broke through’. There’s a million membranes to break through. There’s a million places to go. You know… You move to another direction, another dimension? Big deal! We went to the Moon? Big deal! We went to Mars? Big deal! We keep movin’ and movin’ and movin’. You know… Mohammed went through seven heavens? Big deal, I wanna see the eight heaven! Tenth heaven. Thousandth heaven. You know, it’s like… Break on through to the other side, it’s just like goin’ through one door. One door isn’t enough. A million doors aren’t enough. You have to go beyond… Beyond one reflection. Beyond the mirror. Beyond beyond.
VII
Table of Contents
Abbreviations ... ...IX Summary... ... XIII Sumário... ... XV Chapter 1 Aims and Outline ... 1 Chapter 2 General Introduction ... 5 Chapter 3 Deciphering Protein Arginine Methylation in Mammals ... 15
in Methylation - From DNA, RNA and Histones to Diseases and Treatment, Prof. Anica Dricu (Ed.), ISBN: 978-953-51-0881-8, InTech, DOI: 10.5772/51984 (2012)
Chapter 4 Protein Arginine Methylation Is More Prone to Inhibition by S-Adenosylhomocysteine than DNA Methylation in Vascular Endothelial Cells ... 37 Plos One (2013) 8: e55483
Chapter 5 Folinic Acid Increases Cellular Methylation Capacity and Protein Arginine Methylation in Cultured Human Endothelial Cells ... 53 Submitted
Chapter 6 Global Protein and Histone Arginine Methylation are Affected in a Tissue-Specific Manner in a Rat Model of Diet-Induced Hyperhomocysteinemia ... 61 Biochim. Biophys. Acta (2013) 1832: 1708-1714
Chapter 7 Protein Arginine Hypomethylation in a Mouse Model of Cystathionine Beta-Synthase Deficiency ... 79 Submitted
Chapter 8 Elevation of Liver and Brain Guanidinoacetate in a Mouse Model of Cystathionine Beta-synthase Deficiency ... 97 Submitted
Chapter 9 General Discussion and Perspectives ... 105 List of Publications ... 113 Acknowledgements / Agradecimentos... 115
IX
Abbreviations
Abbreviations ...
Abbreviations
5-mTHF 5-methyltetrahydrofolate
ADMA asymmetric NG,NG-dimethylarginine
AdoHcy S-adenosylhomocysteine AdoMet S-adenosylmethionine AdOx adenosine-2,3-dialdehyde
AGAT L-arginine:glycine amidinotransferase Akt/PKB serine-threonine kinase Akt/protein kinase B
Arg arginine
AZA 5-aza-2′-deoxycytidine
BHMT betaine-homocysteine methyltransferase BORIS brother of the regulator of imprinted sites BTG1 B-cell translocation gene 1 protein BTG2 B-cell translocation gene 2 protein
C cytosine CBP CREB-binding protein CBS cystathionine beta-synthase Cr creatine Ct threshold cycle CTD C-terminal domain Cys cysteine
DDAH dimethylarginine dimethylaminohydrolase DDR DNA damage response
DNMT DNA methyltransferase
eNOS endothelial nitric oxide synthase ERK extracellular signal-regulated kinase ER-α estrogen receptor alpha
EWS Ewing sarcoma FnA folinic acid
FoxO forkhead transcription factor of class O GA Golgi apparatus
GAA guanidinoacetate
GABAAR gamma-aminobutyric acid A receptor GAMT guanidinoacetate methyltransferase GAR glycine- and arginine-rich
GNMT glycine methyltransferase
X
H3R8me2a asymmetrically dimethylated arginine 8 on histone H3 H3R8me2s symmetrically dimethylated arginine 8 on histone H3 H4R3me2a asymmetrically dimethylated arginine 3 on histone H4 H4R3me2s symmetrically dimethylated arginine 3 on histone H4 HAT histone acetyl transferase
hCAF1 chemokine receptor 4-associated factor 1 Hcy homocysteine
HHcy hyperhomocysteinemia HM methionine-enriched
HMLV methionine-enriched, B vitamins-deficient HMT histone methyl transferase
hnRNP heterogeneous nuclear ribonucleoprotein HPLC high-performance liquid chromatography HRE hormone response element
HUVEC human umbilical vein endotelial cells IFN interferon
LC-MS/MS liquid chromatography-tandem mass spectrometry LDH lactate dehydrogenase
L-DOPA L-3,4-dihydroxyphenylalanine LV B vitamins-deficient
MAT methionine adenosyltransferase MBP myelin basic protein
mC 5-methylcytosine Met methionine MMA NG-monomethylarginine MS methionine synthase MTHFR methylenetetrahydrofolate reductase Myr myristoylation NO nitric oxide
NosD nitrous oxidase accessory protein NR nuclear hormone receptor
NUMAC nucleosomal methylation activator complex PAD peptidylarginine deiminase
PC phosphatidylcholine PE phosphatidylethanolamine
PEMT phosphatidylethanolamine methyltransferase PGC-1α proliferator-activated receptor-γ co-activator-1 PKMT protein lysine methyltransferase
XI
Abbreviations
PP2A Ser/Thr protein phosphatase 2A PRMT protein arginine methyltransferase PTM post-translational modification RBP RNA-binding protein
SDMA symmetric NG,N′G-dimethylarginine
SHMT serine hydroxymethyltransferase
STAT signal transducer and activator of transcription tArg total arginine
tC total cytosine tCys total cysteine tHcy total homocysteine THF tetrahydrofolate
THW threonine-histidine-tryptophan TPR tetratricopeptide
XIII
Summary
Summary...
Summary
Homocysteine (Hcy) is a metabolite of the essential amino acid methionine. Elevation of Hcy in plasma, or hyperhomocysteinemia (HHcy), is associated with cardiovascular and neurological diseases by mechanisms still ill-defined. Cellular hypomethylation secondary to build-up of S-adenosylhomocysteine (AdoHcy), the Hcy precursor, may be involved in the pathophysiology of HHcy. AdoHcy is the product of most methylation reactions and, simultaneously, a competitive inhibitor of those reactions.
Our previous investigation has consistently shown that excess AdoHcy can suppress DNA methylation. The working hypothesis for this thesis postulates that, in addition to modulating DNA methylation, AdoHcy may also decrease protein arginine methylation in HHcy. This widespread post-translational modification is emerging as an important player in cell homeostasis.
To test our working hypothesis, we used a cell model in which AdoHcy accumulation was achieved by pharmacological inhibition of AdoHcy production, and two murine models in which the Hcy metabolism was manipulated to promote different patterns of accumulation of Hcy and AdoHcy. Our results documented suppression of global protein arginine methylation induced by AdoHcy accumulation in all studied models. In the cell model, we observed hypomethylation of both DNA and arginine residues in proteins, the effect on proteins being more pronounced that on DNA. In the animal models, decreased arginine methylation was observed in heart and brain of hyperhomocysteinemic rats (moderate HHcy induced by diet), and in liver and brain of mice with severe HHcy due to a genetic defect in Hcy metabolism. Additionally, we were able to document the ability of AdoHcy to decrease histone arginine methylation, a key modulator of gene expression, in the animal model of severe HHcy. Taken together, these studies show that protein arginine hypomethylation may be involved in the mechanistic etiology of disease states associated with HHcy.
We also determined the effect of folate (a nutritional player in Hcy metabolism) treatment and methionine availability on the levels of AdoHcy and protein arginine methylation in our cell model. In fact, this information may be particularly relevant within the context of several recent clinical trials in which folate supplementation reduced Hcy levels, but not cardiovascular disease risk. Our results suggest that Hcy-lowering with folate may be effective in reducing intracellular AdoHcy levels, and therefore in augmenting cellular methylation capacity, only if used in combination with methionine restriction.
Lastly, we focused on one of the most extensive methylation reactions in mammals, the reaction catalyzed by guanidinoacetate methyltransferase. By documenting increased levels of guanidinoacetate in liver and brain of mice with severe HHcy when compared to controls, we reinforce the overall AdoHcy inhibitory effect on methyltransferases activity.
Taken together, our results provide new insights on the pathophysiology of HHcy and lay ground for further studies.
XIV
Keywords: homocysteine; S-adenosylhomocysteine; protein methylation; B vitamins; folic acid; guanidinoacetate
XV
Sumário
Sumário...
Sumário
Níveis elevados da concentração de homocisteína total (tHcy) plasmática, condição designada por hiperhomocisteinémia (HHcy), têm sido associados ao desenvolvimento de patologia cardiovascular e / ou neurológica. Contudo, e apesar da intensa investigação que tem sido feita nesta área, os mecanismos subjacentes a esta associação estão ainda mal definidos. Diversos estudos sugerem que o precursor da homocisteína (Hcy), a S-adenosilhomocisteína (AdoHcy), também se acumula nos estados de HHcy, podendo desempenhar um papel determinante no processo mecanístico subjacente à patologia associada à HHcy devido à sua ação inibitória sobre a atividade das metiltransferases. Estas enzimas usam a S-adenosilmetionina (AdoMet), o precursor da AdoHcy, como dador do grupo metilo, e, como substrato, moléculas de DNA e de proteínas, entre outras. As metiltransferases são responsáveis pela maioria das reações de metilação que ocorrem na célula. Assim, a hipometilação celular secundária à acumulação de AdoHcy poderá explicar o risco de doença associado à HHcy.
A nossa investigação tem mostrado consistentemente que a acumulação de AdoHcy pode suprimir a metilação do DNA. A metilação proteica em resíduos de arginina é uma modificação pós-traducional que participa numa infinidade de processos celulares cruciais e que resulta na formação de resíduos de metilargininas, nomeadamente: NG-monometilarginina (MMA), NG,NG-dimetilarginina assimétrica
(ADMA) e NG,N′G-dimetilarginina simétrica (SDMA). A hipótese de trabalho para esta tese postula
que, para além de afetar a metilação do DNA, a AdoHcy pode igualmente diminuir a metilação dos resíduos de arginina nas proteínas num contexto de HHcy.
O Capítulo 1 da presente tese apresenta os principais objetivos de trabalho e o delineamento dos restantes capítulos. O Capítulo 2 incide sobre aspetos gerais do metabolismo da Hcy, bem como sobre os fatores genéticos e não-genéticos que determinam a HHcy. É ainda fundamentada a hipótese da hipometilação celular como mecanismo patofisiológico na HHcy. No Capítulo 3 é feita uma revisão detalhada subordinada ao tema da metilação proteica em resíduos de arginina, à luz dos conhecimentos mais atuais.
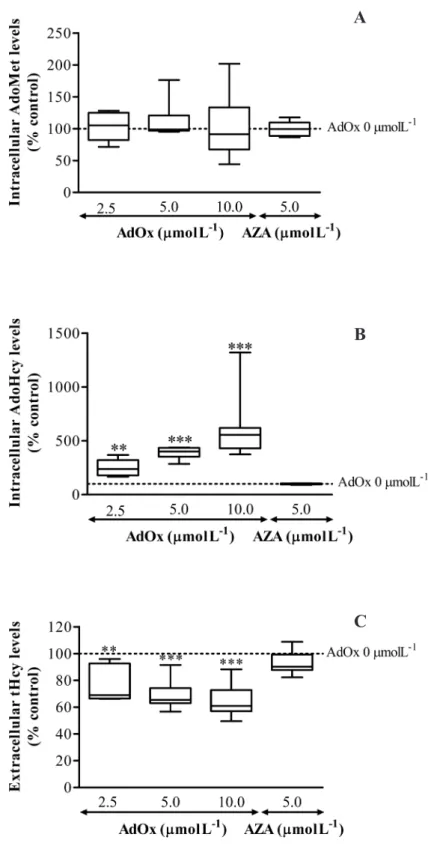
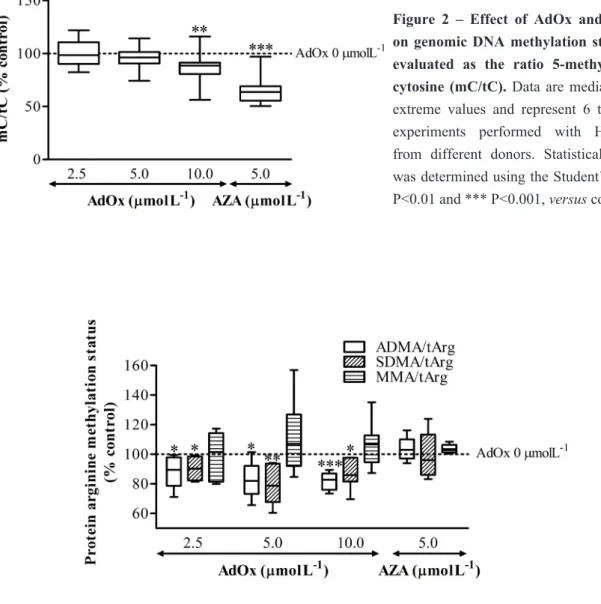
Numerosos estudos têm abordado o envolvimento da perturbação da metilação do DNA em distúrbios do metabolismo da Hcy, ao passo que outras reações de metilação têm sido negligenciadas. O objetivo do estudo apresentado no Capítulo 4 foi o de comparar o impacto da acumulação de AdoHcy na metilação proteica global em resíduos de arginina com o impacto na metilação global do DNA. As células endoteliais da veia umbilical humana (HUVEC) foram usadas como modelo de trabalho, devido à semelhança com o endotélio vascular adulto. A acumulação de AdoHcy intracelular foi obtida através de inibição farmacológica da enzima responsável pela hidrólise de AdoHcy, a AdoHcy hydrolase. Foi observada uma diminuição de cerca de 10% dos teores de ADMA e SDMA incorporados em proteínas, para um aumento de 2.5 vezes da concentração de AdoHcy intracelular. Contudo, a metilação global do DNA apenas sofreu uma diminuição de 10% para um valor de concentração de AdoHcy intracelular cerca de 6 vezes superior à do seu valor basal. Concluímos então que a metilação proteica em resíduos de arginina é mais sensível à acumulação de AdoHcy do
XVI
que a metilação do DNA.
Com o intuito de se diminuir os níveis de tHcy circulante e desta forma se reduzir o risco associado de doença cardiovascular, a terapia com ácido fólico tem sido alvo de diversos ensaios clínicos. Contudo, apesar de se ter confirmado o seu benefício na redução dos níveis de tHcy circulante, a associação com a prevenção da patologia cardiovascular não foi claramente comprovada. A fim de determinar o efeito do ácido fólico nas concentrações dos metabolitos intermediários do metabolismo da Hcy, nomeadamente AdoMet e AdoHcy, incubámos HUVEC com concentrações crescentes de ácido folínico, uma forma de folato. Esta suplementação foi feita em meio de incubação com um teor baixo ou alto de metionina (Capítulo 5). O ácido folínico reduziu a concentração de AdoHcy intracelular e aumentou a monometilação em resíduos de arginina, mas apenas em células incubadas em meio com baixo teor de metionina. Esta observação sugere que a terapêutica com ácido fólico só será eficaz na redução dos níveis intracelulares de AdoHcy e, portanto, no aumento da capacidade de metilação celular, se combinada com uma restrição no teor de metionina.
A fim de comprovarmos se a metilação proteica global em resíduos de arginina era afetada num contexto de HHcy induzida por manipulação da dieta, recorremos ao uso de um modelo animal (Capítulo 6). Com essa finalidade, ratos Wistar foram alimentados com as seguintes dietas: dieta padronizada (controlo); dieta enriquecida em metionina (HM); dieta deficiente em vitaminas B (LV); dieta enriquecida em metionina e deficiente em vitaminas B (HMLV). Qualquer das dietas induziu HHcy, tendo sido mais pronunciado o efeito da dieta HMLV. O aumento do teor de AdoHcy foi observado no fígado com a dieta LV, e no fígado e coração com a dieta HLMV. Nestes dois grupos, observou-se ainda uma diminuição do teor médio de ADMA incorporado em proteínas no cérebro e no coração, em relação ao grupo controlo. Contudo, no tecido cerebral de ratos alimentados com as dietas LV e HMLV, os teores de SDMA e MMA incorporados nas proteínas foram superiores aos do grupo controlo. Neste estudo, analisámos também a metilação de histonas em resíduos de arginina por uma técnica de Western blotting. Observou-se uma diminuição da dimetilação assimétrica no resíduo de arginina 8 na histona H3 (H3R8me2a) no cérebro de ratos alimentados com a dieta HMLV, relativamente ao grupo controlo. Concluímos assim que, num contexto de HHcy induzida por dieta, a metilação proteica global em resíduos de arginina e a metilação da histona H3 no resíduo de arginina 8 sofrem alteração, com especificidade tecidular.
No estudo descrito no Capítulo 7, num modelo de ratinho deficiente em cistationina beta-sintase (CBS), avaliou-se, em diversos tecidos, os teores dos metabolitos intermediários da Hcy, bem como a metilação global do DNA e dos resíduos de arginina proteicos. Os ratinhos com deficiência em CBS apresentaram níveis de Hcy e AdoHcy no fígado, cérebro, coração e rim muito superiores aos encontrados nos ratinhos controlo. Relativamente à metilação global do DNA, não se registaram diferenças entre grupos. No entanto, os ratinhos com deficiência em CBS apresentaram hipometilação proteica em resíduos de arginina no fígado e cérebro, relativamente aos ratinhos controlo. Observou-se também uma diminuição do grau de dimetilação assimétrica no resíduo de arginina 3 na histona H4 (H4R3me2a) no fígado de ratinhos com deficiência em CBS, relativamente aos controlos. Estas
XVII
Sumário
observações sugerem que, na deficiência em CBS, ocorre hipometilação proteica em resíduos de arginina e hipometilação da histona H4 no resíduo de arginina 3, ainda que com especificidade tecidular.
A enzima guanidinoacetato metiltransferase (GAMT) é necessária para a síntese de creatina, sendo inibida pela AdoHcy. Em virtude da elevação dos níveis de AdoHcy observada no modelo de deficiência em CBS estudado no Capítulo 7, no Capítulo 8 pretendemos determinar se neste modelo os níveis de creatina e guanidinoacetato no fígado, cérebro, coração e rim estavam alterados. Observou-se uma elevação significativa do teor de guanidinoacetato no fígado e cérebro de ratinhos com deficiência em CBS, relativamente aos ratinhos controlo. Contudo, os níveis de creatina não sofreram alteração. A toxicidade atribuída ao guanidinoacetato por ativação de recetores do ácido gama-aminobutírico, que tem sido indicada como um dos possíveis mecanismos que contribuem para os sintomas observados na deficiência em GAMT, pode contribuir para o desenvolvimento dos sintomas neurológicos observados na deficiência em CBS.
No Capítulo 9 desta tese é apresentada uma discussão geral dos resultados e das conclusões parcelares de uma forma integrativa, assim como perspetivas para trabalho futuro.
Em conclusão, nesta tese apresentamos um conjunto sólido de evidências, obtidas através da utilização de vários modelos de estudo, que validam a nossa hipótese de trabalho de que a acumulação de AdoHcy se traduz num estado de hipometilação celular. Adicionalmente, mostramos que a metilação proteica em resíduos de arginina pode ser modulada pela dieta. Deste modo, a hipometilação proteica em resíduos de arginina surge como um novo fator a ter em conta no estudo da patofisiologia da HHcy.
Palavras-chave: homocisteína; S-adenosilhomocisteína; metilação proteica; vitaminas B; ácido fólico; guanidinoacetato
CHAPTER 1
Aims and Outline
3
Aims and outline
The association between elevated plasma homocysteine (Hcy) levels and high risk of cardiovascular and neurological diseases in humans has been extensively documented, but the mechanisms at the root of this association remain obscure. Cellular hypomethylation secondary to accumulation of S-adenosylhomocysteine (AdoHcy), the Hcy precursor and an endogenous inhibitor of cellular methylation reactions, may be involved in the pathophysiology of hyperhomocysteinemia (HHcy). This hypothesis has been explored over the past decade, with emphasis given to DNA methylation, but other transmethylation reactions have been dismissed. Protein arginine methylation is a common post-translational modification that is crucially involved in a myriad of cellular events.
The main aims of the present thesis were:
1. to determine whether protein arginine methylation is affected in HHcy;
2. to ascertain the interplay between nutritional status, namely B vitamins availability, and protein arginine methylation status.
Chapter 2 consists in a general introduction to the subjects covered throughout the thesis. It begins with a brief synopsis on the Hcy metabolism and its relationship with human disease. Subsequently, the most common causes of HHcy are described. The working hypothesis that drives the research performed by our group in the Hcy field, namely that cellular hypomethylation may explain Hcy toxicity, is also presented, together with some supporting evidences. Lastly, the negative outcome of recent clinical trials of Hcy-lowering with B vitamins to reduce vascular events is discussed. In Chapter 3, the state of the art of protein arginine methylation is presented. It discloses fundamental information concerning the enzymes responsible for this common post-translational modification, as well as the physiological roles of protein arginine methylation and its regulation. Lastly, the relationship between protein arginine methylation and the Hcy metabolism is discussed.
The involvement of DNA hypomethylation in HHcy has been extensively documented, but, to date, no studies have addressed the hypomethylating effect on protein arginine methylation. Importantly, the enzymes that catalyze this post-translation modification are subject to inhibition by AdoHcy, as are DNA methyltransferases. In Chapter 4, we use an endothelial cell model to investigate whether protein arginine methylation status is affected by AdoHcy accumulation, and the extent of this effect is compared with the hypomethylation effect on DNA.
Hcy-lowering therapy with B vitamins, including folate, has not yield the anticipated cardioprotective effect. However, the effect of folic acid supplementation on the levels of AdoHcy, as well as on cellular methylation processes such as protein arginine methylation, is not known. In Chapter 5, the influence of folate and methionine status on cellular methylation capacity and protein arginine methylation status is examined using the endothelial cell model.
4
Chapter 1
From this point forward, and to test our working hypothesis in vivo, we use two different animal models of HHcy.
In Chapter 6, we investigate how the contents of Met and B vitamins in diet affect cellular methylation capacity and protein arginine methylation status using Wistar rats, in which HHcy is induced by nutritional manipulation (Met enrichment, B vitamins deficiency, or both). Furthermore, the influence of diet manipulation on histone arginine methylation in this animal model is also investigated.
Cystathionine beta-synthase (CBS) deficiency is a rare inborn error of Hcy metabolism that results in severe HHcy. The aim of the study reported in Chapter 7 is to characterize the alterations in the Hcy metabolites in a mouse model of CBS deficiency, and to relate these changes with DNA, histone arginine and global protein arginine methylation status.
Some of the neurological complications observed in CBS deficiency patients are also present in guanidinoacetate methyltransferase (GAMT) deficiency. GAMT catalyzes one of the most extensive methylation reactions in humans, forming creatine (Cr), and is inhibited in vitro by AdoHcy. In Chapter 8, we investigate whether the mouse model used in Chapter 7 presents altered levels of guanidinoacetate (GAA) and Cr.
Lastly, Chapter 9 comprises a general discussion of the results presented in this thesis, as well as perspectives for future work.
CHAPTER 2
General Introduction
7
General introduction
1. Homocysteine Metabolism
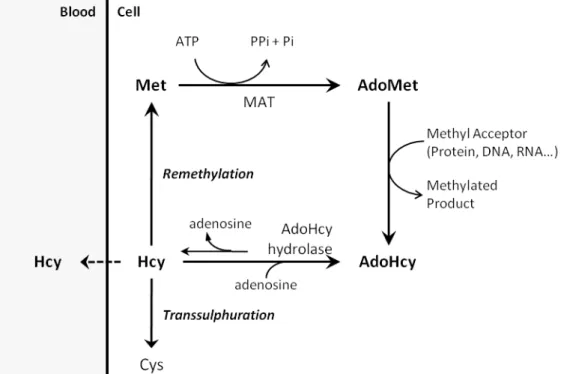
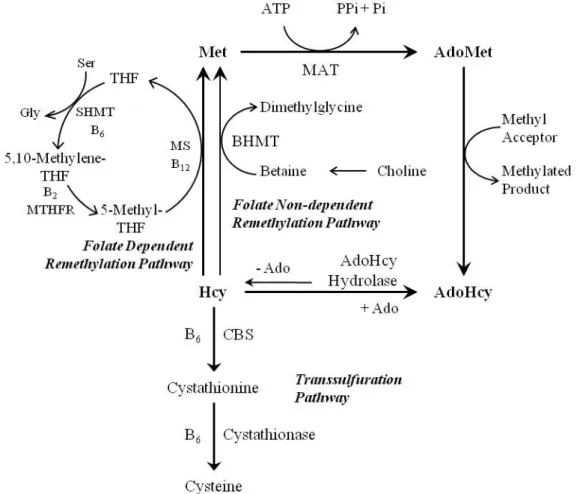
Homocysteine (Hcy) is a sulphur amino acid that circulates in blood mainly in the form of disulfides, either bound to proteins or to other thiol-containing compounds, whereas the reduced form, presenting a free thiol group, only accounts for 1 – 2 % of the total content of Hcy. Total Hcy (tHcy) refers to the sum of all circulating forms of Hcy [1]. Hcy is originated from methionine (Met), which is first condensed with ATP to form S-adenosylmethionine (AdoMet), a common co-substrate involved in methyl group transfers. In fact, AdoMet transfers its methyl group to numerous methyl acceptors, including DNA and proteins [2]. Demethylation of AdoMet yields S-adenosylhomocysteine (AdoHcy), which can be subsequently hydrolyzed to Hcy and adenosine. This reaction, which is reversible, is catalyzed by AdoHcy hydrolase. Importantly, the synthesis of AdoHcy from Hcy and adenosine is thermodynamically favorable, but under normal conditions the reaction proceeds in the hydrolysis reaction due to quick removal of the hydrolysis products. Hcy can be exported or metabolized through the transsulfuration and remethylation pathways. The transsulfuration pathway, in which Hcy is converted to cysteine, relies of the enzymatic activities of vitamin B6-dependent enzymes, namely cystathionine beta-synthase (CBS) and cystathionase. The complete transsulfuration pathway is present only in liver, kidney, small intestine and pancreas [3]. Cysteine is substrate for the rate-limiting step of glutathione synthesis, a major cellular redox buffer. Thus, the transsulfuration pathway plays a key role in controlling the cell’s antioxidant defense system [4]. Additionally, Hcy can be remethylated back to Met through the ubiquitous folate-dependent pathway, where B vitamins are important co-factors, or by betaine-homocysteine methyltransferase (BHMT) activity, which is confined to liver and kidney [3;5].
2. Homocysteine and Disease
In adults, fasting tHcy levels in plasma are usually between 5 and 15 µmolL-1, and tHcy elevation is
classified as moderate (16 – 30 µmolL-1), intermediate (31 – 100 µmolL-1) or severe (>100 µmolL-1)
hyperhomocysteinaemia (HHcy) [6;7]. Meta-analyses have shown that, in the general population, moderate HHcy is an independent and graded risk factor for cardiovascular disease [8-10]. More recently, the spectrum of diseases associated with moderate HHcy was extended to osteoporosis [11] and neurological abnormalities, as Alzheimer’s disease [12], cognitive impairment [13], dementia [14] and schizophrenia [15].
3. Hyperhomocysteinemia in the General Population
Lifestyle factors such as smoking [16], heavy coffee consumption [17] and alcoholism [18] elevate plasma tHcy levels and cause moderate HHcy in the general population. Other causes of moderate HHcy include renal disease [19] and drugs such as methotrexate [20] and valproic acid [21]. Inadequate intake of folate and vitamin B12, involved in the folate-dependent Hcy remethylation
8
Chapter 2
pathway, also promotes tHcy elevation [22]. MTHFR, a crucial enzyme in the folate-dependent Hcy remethylation pathway, catalyzes the reduction of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate (5-mTHF). Then, cobalamin-dependent methionine synthase (MS) catalyzes the transfer of a methyl group from 5-mTHF to Hcy, producing Met and tetrahydrofolate (THF). Serine hydroxymethyltransferase (SHMT) subsequently catalyzes the reversible conversion of serine and THF to glycine and 5,10-methylenetetrahydrofolate. Thus, MTHFR has a crucial role in the recycling of both Met and 5-mTHF. The MTHFR C677T polymorphism yields a thermolabile enzyme with reduced activity in vitro [23]. Homozygosity for the thermolabile MTHFR variant increases plasma tHcy concentrations by approximately 25%, but only in individuals with a low folate status [24].
4. CBS deficiency
CBS deficiency, the most common inborn error of Hcy metabolism, is an autosomal recessive disorder caused by pathogenic mutations in the CBS gene, resulting in severe HHcy and homocystinuria [25]. The biochemical phenotype of CBS deficiency also includes hypocysteinemia and hypermethioninemia, due to increased Hcy remethylation [26]. Individuals with CBS deficiency do not usually display symptoms at birth, but develop serious complications in childhood and throughout life. These may include mental retardation, ectopia lentis, premature thrombotic vascular disease and skeletal abnormalities [27]. Depending on the underlying mutations in the CBS gene, therapy with vitamin B6 (a CBS cofactor) may ameliorate the clinical phenotype [28].
5. Cellular Hypomethylation in Hyperhomocysteinemia
Although the association between Hcy elevation and disease has been known for decades, the underlying mechanisms are still poorly understood. Some of the proposed mechanisms to explain Hcy toxicity are increased oxidative and endoplasmic reticulum stress, lowered nitric oxide availability, protein S-thiolation, N-homocysteinylation and acylation, and cellular hypomethylation (reviewed in [29;30]). The reaction catalyzed by AdoHcy hydrolase is bidirectional and AdoHcy synthesis is thermodynamically favored. Therefore, when Hcy accumulates, AdoHcy will accumulate as well [3]. Notably, AdoHcy is a competitive inhibitor of most AdoMet-dependent methyltransferases [31]. Therefore, the complications associated with HHcy may result from the cellular hypomethylating environment due to AdoHcy-mediated inhibition of methyltransferase activity, a working hypothesis that has been well explored by us during the last decade.
5.1 DNA methylation
DNA methylation is a crucial biological event due to its central role in the regulation of gene expression. In mammals, DNA methylation occurs at cytosine (C) residues that are transformed into
9
General introduction
5-methylcytosine (mC) residues by enzymes termed DNA methyltransferases (DNMTs), which are AdoMet-dependent. DNA methylation is most frequent in CpG islands, which are regions of the genome with a high content of CpG dinucleotides. The deposition of methyl marks within a CpG context, when occurring in a promoter region, is generally associated with transcription suppression [32]. However, DNA methylation does not occur exclusively in a CpG island context [33], is not confined to gene promoters and may also result in transcription activation [34]. Importantly, aberrant DNA methylation patterns have been found in a wide array of diseases, more prominently in cancer [35].
Cumulative evidence supports the notion that Hcy elevation leads to DNA hypomethylation secondary to inhibition of DNMTs by AdoHcy. For instance, we and others have observed that plasma tHcy concentration correlates positively with AdoHcy concentration and negatively with lymphocyte DNA methylation status in healthy individuals and patients with vascular disease [36;37]. Furthermore, a significant reduction in the genomic mC content was detected in advanced human atherosclerotic lesions [38] and in vascular lesions of mice lacking apolipoprotein E, a well-established animal model of atherosclerosis [39]. Studies with murine models of MTHFR and CBS deficiencies have also provided valuable insights into the relationship between Hcy and AdoHcy levels and DNA methylation status. For example, mice with targeted knockout of MTHFR display global DNA hypomethylation in brain and ovaries, along with increased AdoHcy levels in these tissues [40], and mice heterozygous for CBS deficiency display high hepatic levels of AdoHcy and global DNA hypomethylation in liver and testis [41;42].
5.2 Protein methylation
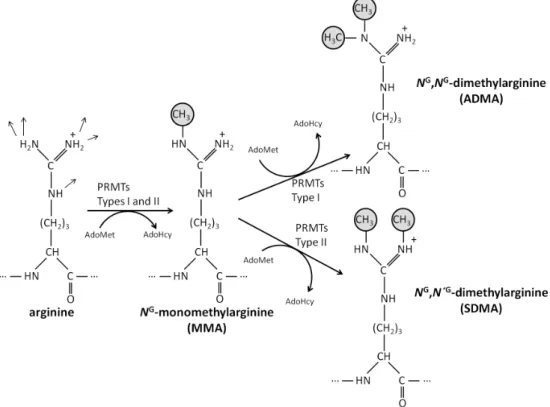
Protein methylation is a common post-translational modification (PTM) that modulates many cellular processes. Several amino acid residues may be methylated (histidine, glutamate, glutamine, asparagine, aspartate, cysteine, and N-terminal and C-terminal residues [43]), but lysine and arginine N-methylation are the most prevalent in mammals. Lysine N-methylation is established by protein lysine methyltransferases (PKMTs) that catalyze the transfer of one to three methyl groups from AdoMet to the lysine ɛ-amine side chain, producing monomethyl-, dimethyl- or trimethyl-lysine residues. In the case of N-methylation of arginine residues in proteins, this PTM is catalyzed by protein arginine methyltransferases (PRMTs), which also rely on AdoMet as methyl group donor. In mammals, there are two types of PRMTs: I and II. All PRMTs form NG-monomethylarginine
(MMA), but whereas type I PRMTs catalyze the formation of asymmetric NG,NG-dimethylarginine
(ADMA) residues, type II PRMTs yield symmetric NG,N´G-dimethylarginine (SDMA) residues. A
detailed review of protein arginine methylation is given in Chapter 3. Histone targeting by PKMTs and PRMTs is involved in chromatin remodeling, protein-nucleosome interactions and transcriptional control of gene expression. However, besides histones, and owing to recent developments in molecular biology and mass spectrometry techniques, many non-histonic targets for PKMTs and PRMTs are being identified. Protein lysine and arginine methylation participates in processes as
10
Chapter 2
diverse as RNA processing, transcription control, protein localization, signal transduction and DNA repair [44;45].
The observations by Perna and colleagues [46] suggested, for the first time, the involvement of protein hypomethylation in disorders of the Hcy metabolism. In this study, AdoHcy-mediated decrease in methyl esterification of erythrocyte membrane proteins was identified in HHcy patients with renal function impairment. In other study, administration of L-DOPA (L-3,4-dihydroxyphenylalanine) to mice reduced AdoMet/AdoHcy ratio and caused hypomethylation of Ser/Thr protein phosphatase 2A (PP2A), a major Tau phosphatase, resulting in increased Tau phosphorylation; folate deficiency was shown to exacerbate this effect [47]. In fact, hypomethylation of PP2A may link Tau hyperphosphorylation, a hallmark in Alzheimer’s disease, with the mildly elevated levels of tHcy and AdoHcy that were observed in patients with Alzheimer’s disease [48]. Moreover, Garcia and colleagues [49] have reported that dietary methyl donor deficiency in rat dams resulted in cardiomyopathy in the offspring, decreased AdoMet/AdoHcy ratio in the myocardium, and impaired arginine methylation of proliferator-activated receptor-γ co-activator-1 (PGC-1α), a transcriptional coactivator that regulates the genes involved in energy metabolism. Additionally, liver steatosis was subsequently identified in these pups due to hypomethylation of PGC-1α in liver and consequent impaired mitochondrial fatty acid oxidation [50].
To the extent of our knowledge, no other studies were conducted to ascertain whether protein methylation decreases in HHcy, and none has focused on global protein arginine methylation.
5.3 Other transmethylation reactions
AdoMet plays a pivotal role as methyl donor in cellular methylation reactions other than DNA and protein methylation. Cumulative evidence has shown that the synthesis of creatine (Cr) by guanidinoacetate methyltransferase (GAMT), of phosphatidylcholine (PC) by phosphatidylethanolamine methyltransferase (PEMT), and of sarcosine by glycine methyltransferase (GNMT), account for the greatest usage of methyl groups from AdoMet [51]. GAMT and PEMT are strongly inhibited by AdoHcy in vitro [52;53]. Accordingly, elevation of guanidinoacetate (GAA) in plasma was found in patients with AdoHcy hydrolase deficiency, who present high levels of AdoHcy [54;55], as well as in patients with combined methylmalonic aciduria and homocystinuria [56]. In a study by Malanovic and colleagues [57], down-regulation of AdoHcy hydrolase in yeast resulted in accumulation of AdoHcy and decreased de novo PC (a major lipid constituent of cell membranes) synthesis due to inhibition of PEMT by AdoHcy. Low concentrations of PC in plasma and erythrocytes were also found in AdoHcy hydrolase deficiency patients [54;55], and rats fed a folate-deficient diet had PC depletion in brain [58]. In contrast to GAMT and PEMT, GNMT is weakly inhibited by AdoHcy. However, this enzyme is thought to play a key role in the regulation of Met and AdoMet levels [59;60]. For instance, the hepatic expression of GNMT is up-regulated in response to dietary Met enrichment in mice [61], whereas GNMT deficiency results in hypermethioninemia [62].
11
General introduction
6. Homocysteine-lowering therapy with B vitamins
Folic acid and other B vitamins play a vital role in Hcy metabolism and cell homeostasis. MS, SHMT and MTHFR are enzymes participating in the folate-dependent remethylation pathway, all using a B vitamin as cofactor (B12, B6 and B2, respectively). In the transsulfuration pathway, vitamin B6 is cofactor of both CBS and cystathionase. Accordingly, administration of B vitamins, including folate, is a well-established therapeutic measure to lower Hcy levels [63]. Therefore, B vitamins supplementation for tHcy reduction should reduce the risk of cardiovascular disease associated with HHcy. To test this possibility, a large number of randomized clinical trials of Hcy-lowering therapy with B vitamins were performed in the past decade, which however did not yielded the anticipated cardio protective effects despite a clear reduction in tHcy levels [63-68]. It is possible that inappropriate trial design has been responsible for such disappointing results. For example, and importantly, patients with levels of tHcy in plasma higher than 20 µmolL-1 were underrepresented
in many of these intervention studies, precluding any definite conclusions regarding the efficacy of treatment for these patients [69].
Other question that remains unanswered is whether administration of B vitamins reduces the levels of intracellular Hcy and metabolites, namely AdoMet and AdoHcy. In addition, excessive folate intake may have adverse effects (e.g. increased inflammation on atherosclerotic lesions) that offset the beneficial effect of Hcy-lowering [69]. As referred above, moderate HHcy has also been associated with neurological abnormalities. Noteworthy, recent results show that Hcy-lowering therapy with B vitamins delays cognitive decline in Alzheimer’s disease [70] and improve cognitive function in the elderly population [71;72]. These observations encourage further efforts to ascertain the potential beneficial effects of Hcy-lowering therapy with B vitamins.
References
1. Di Giuseppe D, Ulivelli M, Bartalini S, Battistini S, Cerase A, Passero S, Summa D, Frosali S, Priora R, Margaritis A & Di Simplicio P (2010) Regulation of redox forms of plasma thiols by albumin in multiple sclerosis after fasting and methionine loading test. Amino Acids, 38, 1461-1471.
2. Grillo MA & Colombatto S (2008) S-adenosylmethionine and its products. Amino Acids, 34, 187-193. 3. Finkelstein JD (1990) Methionine metabolism in mammals. J. Nutr. Biochem., 1, 228-237.
4. Deplancke B & Gaskins HR (2002) Redox control of the transsulfuration and glutathione biosynthesis pathways.
Curr. Opin. Clin. Nutr. Metab Care, 5, 85-92.
5. Sunden SL, Renduchintala MS, Park EI, Miklasz SD & Garrow TA (1997) Betaine-homocysteine methyltransferase expression in porcine and human tissues and chromosomal localization of the human gene. Arch. Biochem.
Biophys., 345, 171-174.
6. Refsum H, Fiskerstrand T, Guttormsen AB & Ueland PM (1997) Assessment of homocysteine status. J. Inherit.
Metab Dis., 20, 286-294.
7. Still RA & McDowell IF (1998) ACP Broadsheet No 152: March 1998. Clinical implications of plasma homocysteine measurement in cardiovascular disease. J. Clin. Pathol., 51, 183-188.
8. Blom HJ & Smulders Y (2011) Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects. J. Inherit. Metab Dis., 34, 75-81.
9. Danesh J & Lewington S (1998) Plasma homocysteine and coronary heart disease: systematic review of published epidemiological studies. J. Cardiovasc. Risk, 5, 229-232.
12
Chapter 2
10. Wald DS, Law M & Morris JK (2002) Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. BMJ, 325, 1202.
11. van Meurs JB, Dhonukshe-Rutten RA, Pluijm SM, van der Klift M, de JR, Lindemans J, de Groot LC, Hofman A, Witteman JC, van Leeuwen JP, Breteler MM, Lips P, Pols HA & Uitterlinden AG (2004) Homocysteine levels and the risk of osteoporotic fracture. N. Engl. J. Med., 350, 2033-2041.
12. Ravaglia G, Forti P, Maioli F, Martelli M, Servadei L, Brunetti N, Porcellini E & Licastro F (2005) Homocysteine and folate as risk factors for dementia and Alzheimer disease. Am. J. Clin. Nutr., 82, 636-643.
13. Haan MN, Miller JW, Aiello AE, Whitmer RA, Jagust WJ, Mungas DM, Allen LH & Green R (2007) Homocysteine, B vitamins, and the incidence of dementia and cognitive impairment: results from the Sacramento Area Latino Study on Aging. Am. J. Clin. Nutr., 85, 511-517.
14. Wald DS, Kasturiratne A & Simmonds M (2011) Serum homocysteine and dementia: meta-analysis of eight cohort studies including 8669 participants. Alzheimers. Dement., 7, 412-417.
15. Muntjewerff JW, Kahn RS, Blom HJ & den HM (2006) Homocysteine, methylenetetrahydrofolate reductase and risk of schizophrenia: a meta-analysis. Mol. Psychiatry, 11, 143-149.
16. de Bree A, Verschuren WM, Blom HJ & Kromhout D (2001) Lifestyle factors and plasma homocysteine concentrations in a general population sample. Am. J. Epidemiol., 154, 150-154.
17. Nygard O, Refsum H, Ueland PM, Stensvold I, Nordrehaug JE, Kvale G & Vollset SE (1997) Coffee consumption and plasma total homocysteine: The Hordaland Homocysteine Study. Am. J. Clin. Nutr., 65, 136-143.
18. Cravo ML, Gloria LM, Selhub J, Nadeau MR, Camilo ME, Resende MP, Cardoso JN, Leitao CN & Mira FC (1996) Hyperhomocysteinemia in chronic alcoholism: correlation with folate, vitamin B-12, and vitamin B-6 status. Am. J. Clin. Nutr., 63, 220-224.
19. Bostom AG & Culleton BF (1999) Hyperhomocysteinemia in chronic renal disease. J. Am. Soc. Nephrol., 10, 891-900.
20. van Ede AE, Laan RF, Blom HJ, Boers GH, Haagsma CJ, Thomas CM, De Boo TM & van de Putte LB (2002) Homocysteine and folate status in methotrexate-treated patients with rheumatoid arthritis. Rheumatology.
(Oxford), 41, 658-665.
21. Ozdemir O, Yakut A, Dinleyici EC, Aydogdu SD, Yarar C & Colak O (2011) Serum asymmetric dimethylarginine (ADMA), homocysteine, vitamin B(12), folate levels, and lipid profiles in epileptic children treated with valproic acid. Eur. J. Pediatr., 170, 873-877.
22. Ubbink JB, Vermaak WJ, van der Merwe A & Becker PJ (1993) Vitamin B-12, vitamin B-6, and folate nutritional status in men with hyperhomocysteinemia. Am. J. Clin. Nutr., 57, 47-53.
23. Yamada K, Chen Z, Rozen R & Matthews RG (2001) Effects of common polymorphisms on the properties of recombinant human methylenetetrahydrofolate reductase. Proc. Natl. Acad. Sci. U. S. A, 98, 14853-14858. 24. Jacques PF, Bostom AG, Williams RR, Ellison RC, Eckfeldt JH, Rosenberg IH, Selhub J & Rozen R (1996)
Relation between folate status, a common mutation in methylenetetrahydrofolate reductase, and plasma homocysteine concentrations. Circulation, 93, 7-9.
25. Gaustadnes M, Rudiger N, Rasmussen K & Ingerslev J (2000) Intermediate and severe hyperhomocysteinemia with thrombosis: a study of genetic determinants. Thromb. Haemost., 83, 554-558.
26. Orendac M, Zeman J, Stabler SP, Allen RH, Kraus JP, Bodamer O, Stockler-Ipsiroglu S, Kvasnicka J & Kozich V (2003) Homocystinuria due to cystathionine beta-synthase deficiency: novel biochemical findings and treatment efficacy. J. Inherit. Metab Dis., 26, 761-773.
27. Skovby F, Gaustadnes M & Mudd SH (2010) A revisit to the natural history of homocystinuria due to cystathionine beta-synthase deficiency. Mol. Genet. Metab, 99, 1-3.
28. Hu FL, Gu Z, Kozich V, Kraus JP, Ramesh V & Shih VE (1993) Molecular basis of cystathionine beta-synthase deficiency in pyridoxine responsive and nonresponsive homocystinuria. Hum. Mol. Genet., 2, 1857-1860. 29. Lentz SR (2005) Mechanisms of homocysteine-induced atherothrombosis. J. Thromb. Haemost., 3, 1646-1654. 30. Perna AF, Ingrosso D, Lombardi C, Acanfora F, Satta E, Cesare CM, Violetti E, Romano MM & De Santo NG
(2003) Possible mechanisms of homocysteine toxicity. Kidney Int. Suppl, S137-S140.
31. Ueland PM (1982) Pharmacological and biochemical aspects of adenosylhomocysteine and S-adenosylhomocysteine hydrolase. Pharmacol. Rev., 34, 223-253.
32. Bernstein BE, Meissner A & Lander ES (2007) The mammalian epigenome. Cell, 128, 669-681.
33. Han H, Cortez CC, Yang X, Nichols PW, Jones PA & Liang G (2011) DNA methylation directly silences genes with non-CpG island promoters and establishes a nucleosome occupied promoter. Hum. Mol. Genet., 20, 4299-4310.
34. Wu H, Coskun V, Tao J, Xie W, Ge W, Yoshikawa K, Li E, Zhang Y & Sun YE (2010) Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science, 329, 444-448.
13
General introduction
36. Castro R, Rivera I, Struys EA, Jansen EE, Ravasco P, Camilo ME, Blom HJ, Jakobs C & Tavares dA, I (2003) Increased homocysteine and S-adenosylhomocysteine concentrations and DNA hypomethylation in vascular disease. Clin. Chem., 49, 1292-1296.
37. Yi P, Melnyk S, Pogribna M, Pogribny IP, Hine RJ & James SJ (2000) Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. J.
Biol. Chem., 275, 29318-29323.
38. Hiltunen MO, Turunen MP, Hakkinen TP, Rutanen J, Hedman M, Makinen K, Turunen AM, Aalto-Setala K & Yla-Herttuala S (2002) DNA hypomethylation and methyltransferase expression in atherosclerotic lesions. Vasc.
Med., 7, 5-11.
39. Lund G, Andersson L, Lauria M, Lindholm M, Fraga MF, Villar-Garea A, Ballestar E, Esteller M & Zaina S (2004) DNA methylation polymorphisms precede any histological sign of atherosclerosis in mice lacking apolipoprotein E. J. Biol. Chem., 279, 29147-29154.
40. Chen Z, Karaplis AC, Ackerman SL, Pogribny IP, Melnyk S, Lussier-Cacan S, Chen MF, Pai A, John SW, Smith RS, Bottiglieri T, Bagley P, Selhub J, Rudnicki MA, James SJ & Rozen R (2001) Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum. Mol. Genet., 10, 433-443.
41. Choumenkovitch SF, Selhub J, Bagley PJ, Maeda N, Nadeau MR, Smith DE & Choi SW (2002) In the cystathionine beta-synthase knockout mouse, elevations in total plasma homocysteine increase tissue S-adenosylhomocysteine, but responses of S-adenosylmethionine and DNA methylation are tissue specific. J. Nutr., 132, 2157-2160. 42. Caudill MA, Wang JC, Melnyk S, Pogribny IP, Jernigan S, Collins MD, Santos-Guzman J, Swendseid ME,
Cogger EA & James SJ (2001) Intracellular S-adenosylhomocysteine concentrations predict global DNA hypomethylation in tissues of methyl-deficient cystathionine beta-synthase heterozygous mice. J. Nutr., 131, 2811-2818.
43. Clarke SG (2013) Protein methylation at the surface and buried deep: thinking outside the histone box. Trends
Biochem. Sci., 38, 243-252.
44. Huang J & Berger SL (2008) The emerging field of dynamic lysine methylation of non-histone proteins. Curr.
Opin. Genet. Dev., 18, 152-158.
45. Pahlich S, Zakaryan RP & Gehring H (2006) Protein arginine methylation: Cellular functions and methods of analysis. Biochim. Biophys. Acta, 1764, 1890-1903.
46. Perna AF, Ingrosso D, Zappia V, Galletti P, Capasso G & De Santo NG (1993) Enzymatic methyl esterification of erythrocyte membrane proteins is impaired in chronic renal failure. Evidence for high levels of the natural inhibitor S-adenosylhomocysteine. J. Clin. Invest, 91, 2497-2503.
47. Bottiglieri T, Arning E, Wasek B, Nunbhakdi-Craig V, Sontag JM & Sontag E (2012) Acute administration of L-DOPA induces changes in methylation metabolites, reduced protein phosphatase 2A methylation, and hyperphosphorylation of Tau protein in mouse brain. J. Neurosci., 32, 9173-9181.
48. Sontag JM, Nunbhakdi-Craig V & Sontag E (2013) Leucine Carboxyl Methyltransferase 1 (LCMT1)-dependent Methylation Regulates the Association of Protein Phosphatase 2A and Tau Protein with Plasma Membrane Microdomains in Neuroblastoma Cells. J. Biol. Chem., 288, 27396-27405.
49. Garcia MM, Gueant-Rodriguez RM, Pooya S, Brachet P, Alberto JM, Jeannesson E, Maskali F, Gueguen N, Marie PY, Lacolley P, Herrmann M, Juilliere Y, Malthiery Y & Gueant JL (2011) Methyl donor deficiency induces cardiomyopathy through altered methylation/acetylation of PGC-1alpha by PRMT1 and SIRT1. J.
Pathol., 225, 324-335.
50. Pooya S, Blaise S, Moreno GM, Giudicelli J, Alberto JM, Gueant-Rodriguez RM, Jeannesson E, Gueguen N, Bressenot A, Nicolas B, Malthiery Y, Daval JL, Peyrin-Biroulet L, Bronowicki JP & Gueant JL (2012) Methyl donor deficiency impairs fatty acid oxidation through PGC-1alpha hypomethylation and decreased ER-alpha, ERR-alpha, and HNF-4alpha in the rat liver. J. Hepatol., 57, 344-351.
51. Mudd SH, Brosnan JT, Brosnan ME, Jacobs RL, Stabler SP, Allen RH, Vance DE & Wagner C (2007) Methyl balance and transmethylation fluxes in humans. Am. J. Clin. Nutr., 85, 19-25.
52. Hoffman DR, Haning JA & Cornatzer WE (1981) Microsomal phosphatidylethanolamine methyltransferase: inhibition by S-adenosylhomocysteine. Lipids, 16, 561-567.
53. Verhoeven NM, Roos B, Struys EA, Salomons GS, van der Knaap MS & Jakobs C (2004) Enzyme assay for diagnosis of guanidinoacetate methyltransferase deficiency. Clin. Chem., 50, 441-443.
54. Buist NR, Glenn B, Vugrek O, Wagner C, Stabler S, Allen RH, Pogribny I, Schulze A, Zeisel SH, Baric I & Mudd SH (2006) S-adenosylhomocysteine hydrolase deficiency in a 26-year-old man. J. Inherit. Metab Dis., 29, 538-545.
14
Chapter 2
55. Baric I, Fumic K, Glenn B, Cuk M, Schulze A, Finkelstein JD, James SJ, Mejaski-Bosnjak V, Pazanin L, Pogribny IP, Rados M, Sarnavka V, Scukanec-Spoljar M, Allen RH, Stabler S, Uzelac L, Vugrek O, Wagner C, Zeisel S & Mudd SH (2004) S-adenosylhomocysteine hydrolase deficiency in a human: a genetic disorder of methionine metabolism. Proc. Natl. Acad. Sci. U. S. A, 101, 4234-4239.
56. Bodamer OA, Sahoo T, Beaudet AL, O’Brien WE, Bottiglieri T, Stockler-Ipsiroglu S, Wagner C & Scaglia F (2005) Creatine metabolism in combined methylmalonic aciduria and homocystinuria. Ann. Neurol., 57, 557-560.
57. Malanovic N, Streith I, Wolinski H, Rechberger G, Kohlwein SD & Tehlivets O (2008) S-adenosyl-L-homocysteine hydrolase, key enzyme of methylation metabolism, regulates phosphatidylcholine synthesis and triacylglycerol homeostasis in yeast: implications for homocysteine as a risk factor of atherosclerosis. J. Biol.
Chem., 283, 23989-23999.
58. Troen AM, Chao WH, Crivello NA, D’Anci KE, Shukitt-Hale B, Smith DE, Selhub J & Rosenberg IH (2008) Cognitive impairment in folate-deficient rats corresponds to depleted brain phosphatidylcholine and is prevented by dietary methionine without lowering plasma homocysteine. J. Nutr., 138, 2502-2509.
59. Takata Y, Huang Y, Komoto J, Yamada T, Konishi K, Ogawa H, Gomi T, Fujioka M & Takusagawa F (2003) Catalytic mechanism of glycine N-methyltransferase. Biochemistry, 42, 8394-8402.
60. Luka Z, Mudd SH & Wagner C (2009) Glycine N-methyltransferase and regulation of S-adenosylmethionine levels. J. Biol. Chem., 284, 22507-22511.
61. DiBello PM, Dayal S, Kaveti S, Zhang D, Kinter M, Lentz SR & Jacobsen DW (2010) The nutrigenetics of hyperhomocysteinemia: quantitative proteomics reveals differences in the methionine cycle enzymes of gene-induced versus diet-gene-induced hyperhomocysteinemia. Mol. Cell Proteomics., 9, 471-485.
62. Augoustides-Savvopoulou P, Luka Z, Karyda S, Stabler SP, Allen RH, Patsiaoura K, Wagner C & Mudd SH (2003) Glycine N -methyltransferase deficiency: a new patient with a novel mutation. J. Inherit. Metab Dis., 26, 745-759.
63. Lonn E, Yusuf S, Arnold MJ, Sheridan P, Pogue J, Micks M, McQueen MJ, Probstfield J, Fodor G, Held C & Genest J, Jr. (2006) Homocysteine lowering with folic acid and B vitamins in vascular disease. N. Engl. J. Med.,
354, 1567-1577.
64. Pan Y, Guo LL, Cai LL, Zhu XJ, Shu JL, Liu XL & Jin HM (2012) Homocysteine-lowering therapy does not lead to reduction in cardiovascular outcomes in chronic kidney disease patients: a meta-analysis of randomised, controlled trials. Br. J. Nutr., 108, 400-407.
65. Khandanpour N, Loke YK, Meyer FJ, Jennings B & Armon MP (2009) Homocysteine and peripheral arterial disease: systematic review and meta-analysis. Eur. J. Vasc. Endovasc. Surg., 38, 316-322.
66. Toole JF, Malinow MR, Chambless LE, Spence JD, Pettigrew LC, Howard VJ, Sides EG, Wang CH & Stampfer M (2004) Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: the Vitamin Intervention for Stroke Prevention (VISP) randomized controlled trial. JAMA,
291, 565-575.
67. Bonaa KH, Njolstad I, Ueland PM, Schirmer H, Tverdal A, Steigen T, Wang H, Nordrehaug JE, Arnesen E & Rasmussen K (2006) Homocysteine lowering and cardiovascular events after acute myocardial infarction. N.
Engl. J. Med., 354, 1578-1588.
68. Bazzano LA, Reynolds K, Holder KN & He J (2006) Effect of folic acid supplementation on risk of cardiovascular diseases: a meta-analysis of randomized controlled trials. JAMA, 296, 2720-2726.
69. Smulders YM & Blom HJ (2011) The homocysteine controversy. J. Inherit. Metab Dis., 34, 93-99.
70. Douaud G, Refsum H, de Jager CA, Jacoby R, Nichols TE, Smith SM & Smith AD (2013) Preventing Alzheimer’s disease-related gray matter atrophy by B-vitamin treatment. Proc. Natl. Acad. Sci. U. S. A, 110, 9523-9528. 71. Durga J, Van Boxtel MP, Schouten EG, Kok FJ, Jolles J, Katan MB & Verhoef P (2007) Effect of 3-year
folic acid supplementation on cognitive function in older adults in the FACIT trial: a randomised, double blind, controlled trial. Lancet, 369, 208-216.
72. Walker JG, Batterham PJ, Mackinnon AJ, Jorm AF, Hickie I, Fenech M, Kljakovic M, Crisp D & Christensen H (2012) Oral folic acid and vitamin B-12 supplementation to prevent cognitive decline in community-dwelling older adults with depressive symptoms--the Beyond Ageing Project: a randomized controlled trial. Am. J. Clin.
CHAPTER 3
Deciphering Protein Arginine
Methylation in Mammals
in Methylation - From DNA, RNA and Histones to Diseases and Treatment, Prof. Anica Dricu (Ed.), ISBN: 978-953-51-0881-8, InTech, DOI: 10.5772/51984 (2012)
Chapter 3 Deciphering Protein Arginine Methylation in Mammals
in Methylation - From DNA, RNA and Histones to Diseases and Treatment, Prof. Anica Dricu (Ed.), ISBN: 978-953-51-0881-8, InTech, DOI: 10.5772/51984 (2012)
17 1. Introduction
The myriad of different post-translation modifications (PTMs) of proteins contributes to the exponential increase of the information encoded by the human genome, from about 25,000 genes to the over 1 million of proteins that composes the human proteome [1;2]. PTMs involve the chemical modification of target amino acid residues and include the addition of a methyl group to arginine residues, or arginine methylation, which is one of the most extensive protein modifications occurring in mammalian cells. During the last years, arginine methylation has gained growing attention due to its impact on cellular function. Attesting to its biological importance, protein arginine methylation
Figure 1 – Methylation of the arginine side chain by protein arginine methyltransferases (PRMTs). The arginine residue holds 5 potential hydrogen bond donors. Mammalian PRMTs
use the methyl group from a molecule of S-adenosylmethionine (AdoMet) to form NG
-monomethylarginine (MMA). Subsequently, type I PRMTs add a methyl group to the same nitrogen atom forming asymmetric NG,NG-dimethylarginine (ADMA), whereas type II PRMT
activity yield symmetric NG,N´G-dimethylarginine (SDMA).
Protein arginine methylation
Deciphering Protein Arginine Methylation in Mammals
Ruben Esse,1,2 Paula Leandro,1,3 Isabel Rivera,1,3 Isabel Tavares de Almeida,1,3 Henk J. Blom,2,4 Rita
Castro1,3
1 Institute for Medicines and Pharmaceutical Sciences (iMed.UL), Faculty of Pharmacy, University of Lisbon, Lisbon,
Portugal
2 Department of Clinical Chemistry, Metabolic Unit, VU University Medical Center, Amsterdam, The Netherlands 3 Department of Biochemistry and Human Biology, Faculty of Pharmacy, University of Lisbon, Portugal
18
Chapter 3
appears to be an ancient modification. Arginine methylation is now recognized as a widespread PTM that occurs on multiple classes of proteins with distinctive cellular localizations. The arginine residue is unique among amino acids, since its guanidine group contains five potential hydrogen bond donors (Fig. 1) positioned for interactions with hydrogen bond acceptors as DNA, RNA and other proteins. Each addition of a methyl group removes a hydrogen donor, so the methylation of arginine residues in proteins will readily modulate their binding interactions and thus their physiological functions. Here, we provide an introduction to protein arginine methylation and discuss the current state of knowledge regarding this modification in mammals. In addition, we provide insight into how protein arginine methylation relates with the homocysteine (Hcy) metabolism. Lastly, we briefly discuss how protein arginine methylation may be disturbed in the context of hyperhomocysteinemia (HHcy), sharing some of our recent results.
2. The mammalian PRMTs: a brief overview
Methylation of arginine is catalyzed by protein arginine methyltransferases (PRMTs). PRMTs catalyze the transfer of methyl groups from the universal methyl donor, S-adenosylmethionine (AdoMet), to the guanidine nitrogen atom of the target arginine residues within proteins [3].
The first documented protein with arginine methylation activity, then termed protein methylase I, was purified from calf thymus [4;5]. Following this discovery, additional family members were identified and purified from different tissues and cell lines [6-16]. PRMTs are grouped in two major types: I and II. Both PRMT types catalyze the transfer of a single methyl group from AdoMet to an arginine residue in the target protein, producing NG-monomethylarginine (MMA) (Fig. 1). However,
differences exist when adding the second methyl group: type I members add it on the same previously
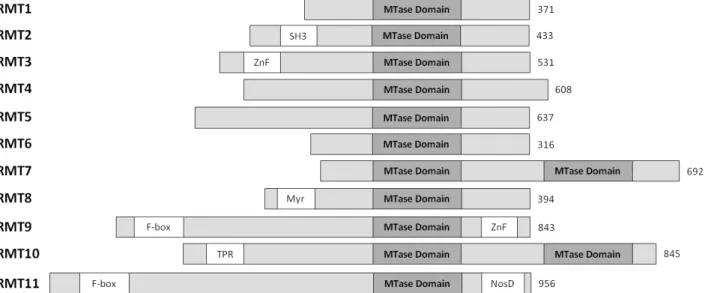
Figure 2 – Schematic representation of the human protein arginine methyltransferase (PRMTs) family. For each
member, the length of the longest isoform is indicated on the right. All members of the family possess at least one conserved MTase (methyltransferase) domain with signature motifs I, post-I, II, and III and a THW loop. Additional domains are marked in yellow boxes: SH3, ZnF (zinc finger), Myr (myristoylation), F-box, TPR (tetratricopeptide) and NosD (nitrous oxidase accessory protein). (Adapted from: [17])
19
Protein arginine methylation
methylated nitrogen, forming NG,NG-dimethylarginine (ADMA), whereas type II members add it to
the other N-terminal nitrogen of the arginine residue, yielding NG,N′G-dimethylarginine (SDMA).
Therefore, three distinct types of methylated arginine residues (MMA, ADMA and SDMA) are present on a horde of different proteins in the cytosol, nucleus and organelles of mammalian cells, ADMA being the most prevalent [3].
To date, the human PRMTs family includes eleven enzymes, termed from PRMT1 to PRMT11. Six are classified as type I (PRMT1, 2, 3, 4, 6, 8), and two as type II (PRMT5, 9). The classification of PRMT7 is controversial, as described below. The remaining 2 proteins, PRMT10 and PRMT11, were identified by their homology with PRMT7 and PRMT9, respectively, but their PRMT activity has not been sustained by experimental evidence [18;19]. The genes encoding the different PRMTs are all located on different chromosomes, except the ones coding for PRMTs 1 and 4, which are both located in chromosome 19. Different splice variants are recognized for all PRMTs. Moreover, different cellular localizations are observed among them.
Although the human PRMT members vary in length from 316 to 956 amino acid residues, they all contain a highly conserved catalytic core region of around 300 residues (Fig. 2). However, each PRMT presents a unique N-terminal region of variable length and distinct domain motifs. For example, PRMT3 and PRMT8 bear a zinc finger (ZnF) and a myristoylation domain, respectively. Interestingly PRMT7 and PRMT10 exhibit a second catalytic domain. Till present, only the 3D structures of type I PRMT1 and 3 and type II PRMT5 were solved by X-ray crystallography [20-22]. These structures revealed that also at the structural level the catalytic core is highly conserved. Three structural regions were identified, namely a methyltransferase (MTase) domain, a β-barrel domain unique to the PRMT family and a dimerization arm. The MTase domain is located in the N-terminal region of the catalytic core. It consists of a α/β Rossmann fold, typical of AdoMet-dependent methyltransferases, containing the conserved motifs I, post-I, II and III [23]. Motif I includes in its C-terminal part the signature sequences for most classes of MTases (GxGxG) involved in AdoMet binding. A relevant structural element localized in the MTase domain is the double-E loop (containing two invariant glutamate (E) residues). The C-terminal part of the catalytic core is a β-barrel domain containing 10 β-strands and a THW (threonine-histidine-tryptophan) loop (the most highly conserved sequence of this domain). Within the overall structure, the THW loop is located next to the double-E loop in the AdoMet binding domain, forming the active site. The dimerization arm is formed by a three-helix segment which is inserted between strands 6 and 7 of the β-barrel domain and is responsible for the dimerization essential for enzyme activity [22].
Most PRMTs methylate arginine residues localized within glycine- and arginine-rich (GAR) sequences but there are exceptions to this rule. For example, PRMT5 and PRMT7 are able to methylate isolated arginine residues [24]. Other factors, such as the accessibility of the target arginine and the conformation of the involved sequence, also govern substrate methylation.
PRMT1 was the first protein with arginine methyltransferase activity in mammalian cells to be cloned [25]. PRMT1, the predominant type I enzyme [26], has been found in all embryonic and adult
20
Chapter 3
tissues examined so far [27]. Due to alternative splicing, there are seven isoforms of the protein, all varying in their N-terminal domain, which are expressed in a tissue-specific manner and have distinct subcellular localization patterns [28]. PRMT1 has a broad substrate specificity with over 40 targets, most being RNA processing proteins, and multiple interacting partners [29]. Targeted Prmt1 knockout in mice results in embryonic lethality [30].
PRMT2 was identified by sequence homology with the rat PRMT1 and its transcripts have been found in most human tissues, with highest levels in heart, prostate, ovary and the neuronal system [10;31]. Only recently, based on the observation that it catalyzes the formation of MMA and ADMA residues on histone H4, PRMT2 was recognized as a type I enzyme [32]. PRMT2 may act in cooperation with PRMT8, since its SH3 domain binds the N-terminal domain of the latter enzyme [33]. In contrast with Prmt1 knockouts, Prmt2−/− mice are viable and grow normally [34]. Some of
the known targets of PRMT2 are STAT3 (signal transducer and activator of transcription 3) [35], histone H4 [32], estrogen receptor alfa [36], the androgen receptor [31], the retinoblastoma gene product [34] and the heterogeneous nuclear ribonucleoprotein (hnRNP) E1B-AP5 [37].
PRMT3, a type I enzyme, was first identified as a PRMT1-binding partner. It is widely expressed in human tissues and has a predominantly cytosolic subcellular localization [16]. PRMT3 possesses a zinc finger domain that assists in its binding to ribosomal proteins, including the S2 protein of the small ribosomal subunit. Although embryos from Prmt3 knockout mice are smaller during development, adults are normal [38].
PRMT4, or CARM1, was first identified as a steroid receptor coactivator [6], providing the first evidence that protein arginine methylation participates in the regulation of gene expression. PRMT4 is a ubiquitously expressed type I PRMT with exclusively nuclear localization that presents restricted substrate specificity. In addition to nuclear hormone receptors, it targets splicing factors [39;40] and ATP-remodeling factors [41]. As opposed to most PRMTs, PRMT4 does not typically methylate GAR sequences. PRMT4 participates in many biological processes, including early T cell development [42], adipocyte differentiation [43], endochondral ossification [44] and proliferation and differentiation of pulmonary epithelial cells [45]. The importance of this enzyme is evidenced by the fact that Prmt4−/− mice are smaller than their wild-type littermates and die perinatally [46].
PRMT5 was the first type II arginine methyltransferase to be acknowledged and is widely expressed in mammals, but at a higher extent in heart, muscle and testis [14]. In contrast with PRMT1, which locates mainly in the nucleus, the human PRMT5 protein is highly expressed in both the nucleus and cytoplasm [47]. Interestingly, PRMT5 localizes to the Golgi apparatus (GA), and loss of PRMT5 disrupts the GA structure, suggesting that PRMT5 functions directly in GA architecture maintenance [47]. In addition, PRMT5 targets a number of histones, transcriptional elongation factors, chromatin remodelers and co-repressors [48], and is critically involved in the maintenance of the spliceosome integrity [49;50]. In mice, loss of PRMT5 results in early embryonic lethality [51].
PRMT6 was identified by sequence homology with other members of the PRMTs family and is localized in the nucleus [8]. PRMT6 is the smallest PRTMs family member (316 amino acids) and