Ciências
Development of purification strategies for SCOMT
and MBCOMT proteins by affinity chromatography
Carla Sofia Ferreira Pereira
Dissertação para obtenção do Grau de Mestre em
Bioquímica
(2º ciclo de estudos)
Orientador: Prof. Doutor Luís António Paulino Passarinha
Coorientadora: Prof. Doutora Ângela Maria Almeida de Sousa
Acknowledgments
Firstly, I would like to express my sincere gratitude to my advisors Professor Luís Passarinha and Professora Ângela Sousa by excellent monitoring and immense knowledge scientific. I have no words for the confidence placed in me and in my work. Thank you for motivation to never give up my objectives. I must mention that it was for me a real privilege to work beside you.
To my dear family, parents and sister, that I love very much, want to thank all the support, unconditional love but also for patience in difficult times and to be present at all times of my life.
I would also like to express my gratitude to Margarida Grilo and Fátima Santos, whose expertise, advice and encouragement made my work much more easy.
I specially thank my lab colleagues for the unconditional help, friendship and support, and all the fun we had along this journey.
I would like to thank the Universidade da Beira Interior and Centro de Investigação em Ciências da Saúde for allowing the development of my research work.
Last but not the least, I would like to thank all my friends, particulary, Cláudia Faria and Leonor Varandas for the encouragement all this time and for helping me make me the person I am today, thank all friendship.
Resumo alargado
A catecol-O-metiltransferase (COMT, CE 2.1.1.6) consiste numa enzima metiltransferase dependente de magnésio que catalisa a metilação de substratos catecóis utilizando S-adenosyl-L-metionina (SAM) com dador do grupo metil, originando dois produtos O-metilados.
Esta enzima encontra-se densamente expressa ao longo do córtex pré-frontal e do sistema límbico. Nos seres humanos, a COMT existe sob a forma de duas isoformas, uma solúvel (SCOMT) localizada no citoplasma e que tem como principal função a eliminação de catecóis biologicamente ativos e, outra associada a membranas plasmáticas (MBCOMT) que desempenha um papel importante no metabolismo in vivo das catecolaminas. Ambas as isoformas são codificadas pelo mesmo gene, localizado no cromossoma 22, a partir de dois promotores, P1 (promotor transcrito a 1.5 kb relativamente à isoforma MBCOMT) e, P2 (promotor transcrito a 1.3 kb em relação à isoforma SCOMT). Ambas são expressas na maioria dos tecidos humanos, exceto no cérebro, onde a MBCOMT é a isoforma dominante.
Em geral, a função fisiológica da COMT é a eliminação de catecóis biologicamente ativos ou tóxicos, funcionando como uma barreira desintoxicante entre o sangue e vários tecidos. Especificamente, a COMT encontra-se envolvida na inativação dos neurotransmissores no sistema nervoso central e regulação dos sistemas dopaminérgicos e noradrenérgicos.
A COMT ao longo dos últimos anos tornou-se um alvo de estudos de diversas patologias, tais como a doença de Parkinson (PD). A PD caracteriza-se pela degeneração neuronal dopaminérgica na substância nigra, induzindo a uma diminuição dos níveis do transmissor da dopamina no cérebro. A degradação dos níveis de dopamina iniciados pela COMT conceberam estratégias de terapia. As atuais terapias passam pela administração de levodopa (precursor da dopamina) conjuntamente com inibidores da COMT e da monoamina oxidase (MAO). Portanto, existe um grande interesse científico no desenvolvimento de estratégias de purificação para estudos estruturais de ambas as isoformas, de modo a se desenhar moléculas capazes de inibir a COMT.
Assim, o principal objetivo deste trabalho consistiu no desenvolvimento de um processo cromatográfico sustentável de forma a obter quantidades significativas da proteína SCOMT_6His na sua forma ativa e pura para estudos cinéticos e estruturais. Deste modo, pela primeira vez, duas estratégias cromatográficas foram propostas para a recuperação de SCOMT_6His a partir de lisados P. pastoris usando o monólito Agmatina depois de uma pré-purificação com a coluna de Q-Sepharose ou diretamente depois de um processo de filtração. Estudos realizados pelo nosso grupo de investigação demonstraram que a aplicação da Q-Sepharose como permutador aniónico revelou ser eficiente na recuperação de ambas as isoformas. No entanto, neste trabalho houve a necessidade de se adaptar o processo cromatográfico. Assim, tornou-se conveniente analisar a aplicação de vários gradientes com o
mostraram que a retenção da proteína SCOMT_6His é alcançada com a aplicação de um aumento linear do gradiente de cloreto de sódio (NaCl) de 0 a 100 mM NaCl seguido de outro gradiente linear de 100 a 310 mM NaCl enquanto, a sua eluição é atingida com um gradiente por passos a alta concentração (450 mM NaCl).
Em termos de recuperação, a coluna Q-Sepharose não demonstrou uma elevada seletividade para o isolamento SCOMT_6His, pois verificou-se uma perda significativa na atividade específica da proteína de interesse em relação à amostra de lisado.
Para melhorar o grau de pureza da proteína alvo obtida pelo ensaio da Q-Sepharose, vários suportes monolíticos foram testados (CDI, Histamina e Agmatina). Os suportes monolíticos têm sido aplicados com sucesso para a purificação de DNA plasmídico, RNA e proteínas, no entanto, estes suportes nunca foram utilizados na purificação da proteína SCOMT_6His. Deste modo, o estudo destes três suportes teve como finalidade avaliar o comportamento cromatográfico da amostra pré-purificada a fim de se explorar diferentes estratégias de eluição por aumento e diminuição de concentrações de cloreto de sódio e sulfato de amónio ((NH4)2SO4). De acordo com os resultados obtidos, as estratégias de eluição da proteína
SCOMT_6His adotada para monólitos CDI e Histamina foram baseados na manipulação das condições hidrofóbicas e iónicas, no entanto, verificou-se que a proteína alvo não ficou retida nestes monólitos, sendo eluída no flowthrough em conjunto com outras proteínas contaminantes. Por sua vez, a retenção da SCOMT_6His foi conseguida no monólito Agmatina e, a sua eluição foi possível com um gradiente linear crescente de NaCl na fase móvel. Por fim, com o objetivo de se isolar e purificar a SCOMT_6His no monólito Agmatina, houve a necessidade de se injetar diretamente a amostra de lisado de P. pastoris no suporte, pois com esta estratégia aumentou-se a quantidade de proteína injetada na matriz originando menores perdas no rendimento do passo cromatográfico.
Em conclusão, a comparação destas abordagens demonstram as limitações existentes no delineamento das estratégias de purificação desta proteína. No entanto, os estudos realizados nos monólitos apoiam claramente que estes têm vantagens sobre os métodos anteriores publicados, devido à sua simplicidade no processo purificação.
Palavras-chave
Parkinson, COMT solúvel, Purificação, Cromatografia de Interação Aniónica, Cromatografia de Afinidade, Suportes monolíticos.
Abstract
Catechol-O-methyltransferase (COMT, EC 2.1.1.6) was first described in 1958. It is a S- adenosyl-L-methionine (SAM) dependent methyltransferase which catalyses the methylation of
catechol substrates (catecholamines, catecholestrogens). This protein plays an important role in the brain, since participates in the metabolism of the neurotransmitter dopamine, being involved in neurodegenerative diseases such as Parkinson's disease. Biosynthesis and purification methods have allowed the crystallization of the soluble COMT (SCOMT) from rats
and the analysis of the kinetic properties of the enzyme in detail. In this study, the main goal was to develop appropriate strategies for purification of SCOMT_6His by using initially the Q-sepharose column to clarify the sample and then monolithic supports such as CarbonylDiImidazole (CDI, Histamine and Agmatine monoliths. Firstly, recombinant SCOMT_6His production was performed using Pichia pastoris X33 cells containing the expression construct pICZα A-hSCOMT_His6. Subsequently, a suitable cell lysis stage employing glass beads was performed, and the lysate was recovered and directly injected onto the Q-sepharose support for clarification and reduction of the homologous proteins from P. pastoris lysates. Results shown that for a complete adsorption of SCOMT_6His onto the anionic resin it was necessary a linear salt gradient (0 mM to 310 mM NaCl in 10 mM Tris-HCl, pH 7.8). Subsequently, for SCOMT_6His elution it was performed a stepwise salt gradient of 450 mM and 1 M of NaCl in 10 mM Tris-HCl, pH 7.8. By analysis of several eluted peaks with SDS–PAGE gel and western blot, it can be observed that SCOMT_6His was eluted essentially at one fraction with high NaCl concentration. Also activity levels and SCOMT_6His recovery rates were evaluated after Q-Sepharose chromatography. Thereafter, the pre-purified sample was injected in three monolithic supports (CDI, Histamine and Agmatine) in order to explore different elution strategies by increasing and decreasing of sodium chloride and ammonium sulphate concentrations. According to the conducted studies, it was found that the SCOMT_6His protein was not retained in CDI and Histamine monoliths under hydrophobic and ionic elution conditions. However, after the equilibrium of the Agmatine monolith with 10 mM Tris–HCl buffer at pH 7.8 at 1 mL/min, the SCOMT_6His was retained, being eluted with 1.5 M NaCl in 10 mM Tris-HCl at pH 7.8. Finally, the direct injection of filtrate lysate sample was also tested in the Agmatine monolith by increasing the NaCl concentration in the elution strategy in order to isolate and purify the SCOMT_6His.
Keywords
Table of Contents
Chapter I ... 1
Introduction ... 1
1.1 The enzyme catechol-O-methyltransferase ... 1
1.1.1 Functions of COMT ... 2
1.1.2 Isoforms: SCOMT and MBCOMT ... 3
1.1.3 Genetic Polymorphisms ... 3
1.1.4 Stability of COMT ... 4
1.1.5 COMT inhibitors ... 5
1.1.6 COMT inhibition in Parkinson`s disease ... 7
1.2 Properties of COMT isoforms ... 7
1.2.1 Biosynthesis of SCOMT and MBCOMT ... 8
1.3 Chromatography ... 9
1.3.1 Chromatographic Matrix ... 10
1.3.2 Chromatographic methods for SCOMT and MBCOMT purification ... 11
1.3.3 Ionic Exchange Chromatography ... 14
1.3.4 Monolithic supports ... 16
Justification and Objectives ... 20
Chapter II ... 21
Materials and Methods ... 21
2.1 Materials ... 21
2.2 Plasmids, bacterial strains and media ... 21
2.3 Recombinant SCOMT_6His production and recuperation... 22
2.4 Pre-purification by Anion Exchange Chromatography ... 23
2.5 Purification by Monoliths ... 23
2.6 Total protein quantification... 24
2.7 SDS-PAGE and Western blot ... 24
2.8 SCOMT_6His enzymatic assay ... 25
Chapter III ... 26
Results and Discussion ... 26
3.1 Production of SCOMT_6His ... 26
3.2 SCOMT_6His recovery assays on Q-Sepharose ... 27
3.3 SCOMT_6His recovery assays on Monoliths ... 36
3.3.1 CarbonylDilmidazole Monolith ... 36
3.3.2 Histamine Monolith ... 40
Chapter IV ... 52 Conclusions ... 52 Chapter V ... 54 Future perspectives ... 54 Chapter VI ... 55 References ... 55 Chapter VII ... 62 Appendices ... 62
List of Figures
Figure I - A typical reaction catalysed by COMT ... 1
Figure II - Representation of the three-dimensional structure of COMT ... 2
Figure III - Representation of some structures of first generation COMT inhibitors ... 5
Figure IV - Representation of some structures of second generation COMT inhibitors ... 6
Figure V - Representation of metabolic pathway of L-Dopa ... 6
Figure VI - Representation of a typical column apply in chromatography ... 9
Figure VII - Representation of the phases of chromatographic procedure ... 10
Figure VIII - Structure of Sepharose ... 11
Figure IX – A typical profile from ion exchange chromatography ... 14
Figure X - Effect of pH on protein binding and elution patterns in Ionic Exchange Chromatography ... 15
Figure XI - Immobilization of ligands by CarbonylDilmidazole monolithic support ... 17
Figure XII - Schematic representation of immobilized histamine ligands ... 18
Figure XIII - Schematic representation of immobilized agmatine ligands... 18
Figure XIV- A typical plasmid growth curve in P. pastoris from SCOMT_6His biosynthesis ... 26
Figure XV - The SCOMT_6His chromatographic profile on Q-Sepharose (1st strategy) ... 29
Figure XVI – SDS-PAGE analysis (A) and Western Bolt (B) of the recovered fractions from the SCOMT_6His chromatographic assay on Q-sepharose of figure XV ... 30
Figure XVII - The SCOMT_6His chromatographic profile on Q-Sepharose (2st strategy) ... 31
Figure XVIII – SDS-PAGE analysis (A) and Western Bolt (B) of the recovered fractions from the SCOMT_6His chromatographic assay on Q-sepharose of figure XVII ... 32
Figure XIX - The SCOMT_6His chromatographic profile on Q-Sepharose (3st strategy) ... 32
Figure XX – SDS-PAGE analysis (A) and Western Bolt (B) of the recovered fractions from the SCOMT_6His chromatographic assay on Q-sepharose of figure XIX ... 34
Figure XXI - Chromatographic profiles obtained by HPLC analysis ... 35
Figure XXII - The SCOMT_6His chromatographic profile on CDI monolith in hydrphobic conditions ... 37
Figure XXIII – SDS-PAGE analysis of the recovered fractions from the SCOMT_6His chromatographic assay on CDI monolith of figure XXII ... 38
Figure XXIV - The SCOMT_6His chromatographic profile on CDI monolith in ionic conditions .. 39
Figure XXV – SDS-PAGE analysis of the recovered fractions from the SCOMT_6His chromatographic assay on CDI monolith of figure XXIV ... 40
Figure XXVI - The SCOMT_6His chromatographic profile on Histamine monolith in hydrphobic conditions ... 41
Figure XXVII – SDS-PAGE analysis of the recovered fractions from the SCOMT_6His chromatographic assay on Histamine monolith of figure XXVI ... 42 Figure XXVIII - The SCOMT_6His chromatographic profile on Histamine monolith in ionic conditions ... 43 Figure XXIX – SDS-PAGE analysis of the recovered fractions from the SCOMT_6His chromatographic assay on Histamine monolith of figure XXVIII ... 44 Figure XXX - The SCOMT_6His chromatographic profile on Agmatine monolith in ionic conditions ... 45 Figure XXXI – SDS-PAGE analysis (A) and Western Bolt (B) of the recovered fractions from the SCOMT_6His chromatographic assay on Agmatine monolith of figure XXX ... 46 Figure XXXII – SDS-PAGE analysis (A) and Western Bolt (B) of the recovered fractions from the pre-treatment of P. pastoris lysate ... 47 Figure XXXIII – The SCOMT_6His chromatographic profile on Agmatine monolith ... 48 Figure XXXIV – SDS-PAGE analysis (A) and (B) from the SCOMT_6His chromatographic assay on Agmatine monolithic of figure XXXIII ... 49 Figure XXXV – The SCOMT_6His chromatographic profile on Agmatine monolith with filtred lystate ... 50 Figure XXXVI – SDS-PAGE analysis (A) and Western Bolt (B) from the SCOMT chromatographic assay on Agmatine monolithic of figure XXXV ... 51
List of Tables
Table I - Types of ion exchangers ... 16 Table II – Adsorption and elution behaviour of SCOMT protein in the Q-sepharose column at NaCl concentrations described by our research group ... 28 Table III – Recombinant SCOMT_6His activity levels after recovery by AEC using Q-Sepharose as anion exchanger. ... 35
List of Acronyms
3,5-DNC 3,5-dinitrocatechol 3-OMD 3-O-methyl-levodopa
AADC Peripheral aromatic L-amino acid decarboxylase
AC Affinity Chromatography AdoMet S-Adenosyl-L-methionine
B.choshinensis Brevibacillus choshinensis
BSA Bovine serum albumin CDI CarbonylDilmidazole
CEA Anionic exchange chromatography CEC Cationic exchange chromatography COMT Catechol-O-metytransferase CV Column volume
DNA Deoxyribonucleic acid
E.coli Escherichia coli
HCl Hydrogen chloride
HIC Hydrophobic Interaction Chromatography His Histidine aminoacid
HPLC High performance liquid chromatography IEC Ion Exchange Chromatography
IMAC Immobilized metal-affinity chromatography KDa kilodaltons
Km Michaelis- Menten constant L-Dopa Levodopa
MAO Monoamine oxidase
MBCOMT Membrane bound catechol-O-methyltransferase Met Methionine Mg2+ Magnesium ion MgCl2 Magnesium chloride MP’s Membrane proteins Lys Lysine OD600 Optical density at 600nm OPC Opicapone PD Parkinson disease PI Isoelectric point
P. pastoris Pichia pastoris
Pro Proline
SAH S-adenosyl-L-homocysteine
SAM S-adenosyl-L-methionine
SCOMT Soluble catechol-O-methyltransferase SDS Sodium dodecyl sulphate
SDS-PAGE Reducing sodium dodecyl sulphate-polyacrylamide gel electrophoresis Tris Tris(hydroxymethyl)aminomethane
Vmax Maximum velocity
Chapter I
Introduction
1.1 The enzyme catechol-O-methyltransferase
Catechol-O-methyltransferase (COMT, E.C.2.1.1.6.) was first characterized by Axelrod and Tomchick in 1958 [1]. Both investigators showed that the enzyme is responsible for the 3-O-methylation of catecholamines [2]. This enzyme was firstly discovered in rat liver extracts, and since then, it has been found in plants, yeast, fungi, invertebrates and vertebrates [3, 5]. In mammals, COMT is widely distributed throughout the organs of the body and their higher activity levels have been found in liver, kidney and gut wall. This enzyme is densely expressed throughout both the prefrontal cortex and the limbic system. It is noteworthy that liver is the most important site for the metabolism of circulating catechol containing molecules [1, 4, 5].
COMT is a monomeric magnesium-dependent enzyme that catalyses the methylation of catechol substrates using S-adenosyl-L-methionine (SAM) as a methyl donor, taking as reaction
products, O-methylated catechol and S-adenosyl-L-homocysteine (SAH) (Figure I) [3, 5, 6, 11,
16, 24].
Figure I - A typical reaction catalysed by COMT. SAM: S-adenosyl-L-methionine; SAH: S-adenosyl-L
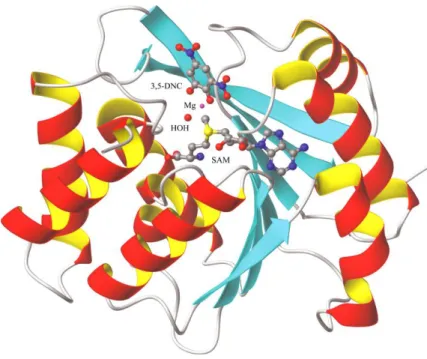
Regarding its three-dimensional structure, the protein is composed of a seven stranded β-sheet, wedged between two sets of α-helices as seen in Figure II [8]. The active site of COMT consists of the S-adenosyl-L-methionine (AdoMet) binding domain and few amino acids such as
Lysine and Proline that are important for the binding of the substrate, water, and Mg2+. It is
noteworthy that the binding motif of the AdoMet site is similar to the Rossman fold [8, 9]. In addition, Mg2+ ions are essential for COMT activity, since they are coordinated to both of the
catecholic hydroxyls, to a water molecule and to three amino acid residues in the catalytic site of COMT [2].
Figure II - Representation of the three-dimensional structure of COMT. The S-adenosyl-L-methionine
co-substrate (SAM), the 3,5-dinitrocatechol (3,5-DNC), the magnesium ion (Mg2+), and coordinated water
molecules are depicted (adapted from [8]).
For example, amino acid residues such as Lys144 (Lysine 144) accept a proton of the hydroxyl group and residues such as Trp38 (Tryptophan 38), Trp143 (Tryptophan 143) and Pro174 (Proline 174) form hydrophobic walls that define COMT selectivity for the substrate [8].
1.1.1 Functions of COMT
Over the recent years COMT has become a target in pharmacological studies due to its interference in normal brain function and possible involvement in some human disorders such as Parkinson disease (PD), Alzheimer disease and schizophrenia due to lack of dopamine [3, 4, 8, 10, 29].
In general, COMT physiological function consists in the inactivation of biologically active or toxic catechols. Specifically, COMT it is involved in the inactivation of the neurotransmitters in the central nervous system and regulation dopaminergic and noradrenergic systems. This protein catalyses the transfer of a methyl group to catecholamines and degrades dopamine, norepinephrine and epinephrine [5, 12]. Beyond these specifications, this protein has important role in the inactivation of catecholamines, metabolism of catecholestrogens and
catecholic drugs, such as L-dopa and carbidopa [2]. Therefore, COMT has been implicated in
several human diseases such as cardiovascular diseases, estrogen induced cancers and neurologic disorders [2, 3].
1.1.2 Isoforms: SCOMT and MBCOMT
In humans, COMT presents two molecular forms, a soluble (SCOMT) and a membrane-bound (MBCOMT) [1, 2, 6].
SCOMT is a nonglycosylated protein containing 221 amino acid residues and a molecular weight of 24.7 kDa [1, 2, 10, 11]. This soluble form presents most abundance in the cytoplasm and has principal function as elimination of biologically active or toxic catechols [5, 11, 28]. Relatively to MBCOMT, it is an integral membrane protein and is found mainly associated with the rough endoplasmic reticulum membrane [2, 10, 11, 13, 28].This isoform has an additional peptide in its amino terminal of 50 amino acid residues and a molecular weight of 30 kDa [2, 5, 28]. This extra peptide contains a stretch of 21 hydrophobic amino acid residues that constitute the membrane anchor region [5, 6, 10]. This enzyme plays an important role of metabolism of catecholamines in vivo since it has 100-fold higher affinity for catecholamine substrates that soluble isoform [6, 13].
Finally, it is important to note that both forms of COMT are coded by a single gene that is located on chromosome 22 and is composed of six exons [4, 8]. The expression of the COMT gene is controlled by two distinct promoters located in exon 3 [9].
1.1.3 Genetic Polymorphisms
COMT presents two polymorphic forms, a thermolabile low activity form contains Met-108 (158 in MB-COMT) and a thermostable high activity form contains Val-108 (158 in MB-COMT) [2]. Moreover, the COMT polymorphism has been shown to account for individual variability in the response to pharmacological manipulations that alter dopamine [20].
In human tissues, COMT activity is distributed to three levels, low (COMTLL), intermediate
(COMTLH), and high (COMTHH) [9]. The most studied genetic polymorphism consists in the
substitution of amino acid valine (Val) by methionine (Met) (Val158Met). This functional polymorphism is caused by transition of guanine to adenine at codon 158 of the MBCOMT and is associated for example to Parkinson's disease [9, 14, 15, 21]. The Met108/158 variant is associated with low enzymatic activity and decreased thermal stability, while the Val108/158 is associated with high activity [14, 15, 20, 21]. However, there are other polymorphisms in the COMT gene, such as Ala22Ser (G/T), His12His (C/T), Leu86Leu (C/G and C/T) and Ala52Thr (G/A) in which COMT activity is not affected by mutation [14, 15].
1.1.4 Stability of COMT
The clinical use of therapeutic proteins has increased over the years due to scientific developments and continued growth of biotechnology and biopharmaceutical industries [8]. Therefore, this strategy has enabled the treatment of a wide range threatening diseases [29]. However, the aggregation and misfolding continues to be a problem in experimental approaches using soluble proteins. The tendency to form aggregates induces a decrease in biological activity and reduces the efficiency of separation techniques [30]. In this context, various purification techniques have been developed in order to increase the activity rate and consequently its performance [29, 30]. In order to avoid the activity losses of therapeutic proteins, there are several parameters to analyse such as pH and ionic strength. The ionic strength should be adjusted to the minimum that allows a homogeneous solubilisation and to a maximum which avoid the dissociation of protein structures [8, 29].
The protein COMT is highly unstable and loses rapidly its activity during isolation and storage [2, 9]. The temperature is one of the factors that lower activity. Other factor is pH, essential to ensure maintenance of catalytic activity and specifically for COMT optimum pH is achieved between 7.5 and 8.0 [27].
The enzyme contains few cysteine residues in its primary structure and a reasonable reason for COMT poor stability is the oxidation of thiol (-SH free groups of cysteine) and the consequent formation of intra- or intermolecular disulphide bridges [31]. According to the literature the cofactor SAM and magnesium chloride (MgCl2) reduce cysteine oxidation,
preventing COMT inactivation, since these residues are essential to catalytic activity [5, 31]. In addition, several types of stabilizers have a stabilizing effect on proteins protecting them against loss of activity and thermal denaturation such as sugars, divalent metals, glycerol and some amino acids (glycine and proline) [32].
1.1.5 COMT inhibitors
The study and development of COMT inhibitors began in 1975 by authoring of Guldberg Marsden [14] and led to an improvement in the treatment of Parkinson’s disease [3, 6, 11, 25].
There are three generations of inhibitors. In the first generation are included derivatives of pyrogallol and catechols, such as gallic acid, caffeic acid, U-0521, 2-hydroxyoestrogens, or flavonoids like quercetin or rutin [9]. Other noncatecholic compounds were also identified such as ascorbic acid, tropolones, and derivatives of 8-hydroxyquinolines and 3-hydroxylated pyrones and pyridines (Figure III) [3, 4, 9]. Typically, inhibitors such as tropolone and pyrogallol present low efficacy in vivo and are toxic.
Figure III - Representation of some structures of first generation COMT inhibitors (adapted from [3]).
The second generation of COMT inhibitors includes nitrocatechols as entacapone (OR-611), nitecapone (OR-462) and tolcapone (Ro 40-7592) (Figure IV) [3, 4, 9] have been extensively studied in the context of PD. [6, 8, 16, 22, 25]. These COMT inhibitors have beneficial effects in increasing the half-life of levodopa (L-Dopa), a drug used as substitute of dopamine (Figure V) [3, 4, 20].
Figure IV - Representation of some structures of second generation COMT inhibitors (adapted from [3]).
Figure V - Representation of metabolic pathway of L-Dopa (adapted from [1]). L-Dopa: Levodopa; DDC:
Dopa decarboxylase; DA: Dopamine; COMT: Catecol-O-metiltransferase; SAM: S-adenosyl-L-methionine;
SAH: S-adenosyl-L-homocysteine 3-OMD: 3-O-metil-Levodopa.
Finally, the third generation COMT inhibitor is opicapone (OPC). OPC is a hydrophilic 1,2,4-oxadiazole analogue with a pyridine N-oxide residue at position 3 providing high COMT inhibitory potency and avoiding cell toxicity [17, 18,19]. Another advantage of this inhibitor
1.1.6 COMT inhibition in Parkinson`s disease
PD is the most common chronic neurodegenerative disease that affects movement behaviour. PD is characterised by dopaminergic neuronal degeneration in the substantia nigra and consecutively by striatal dopamine loss with the accumulation of the protein α-synuclein [4, 17]. This happens because COMT initiates the degradation of brain synaptic dopamine levels by introducing a methyl-group from SAM and then later, methylated dopamine is further degraded by monoamine oxidase (MAO) [18, 21].
The treatment of this disease consists in the dopamine replacement therapy with levodopa together with an inhibitor of aromatic amino acid decarboxylase and a COMT inhibitor [5, 6, 9]. The therapeutic effect of levodopa depends on its biotransformation to dopamine in the brain. However, levodopa undergoes rapid and extensive metabolization by peripheral aromatic L-amino acid decarboxylase (AADC) and COMT [19]. Therefore levodopa is usually
co-administered with an AADC inhibitor (carbidopa or benserazide) which increases levodopa bioavailability, but still approximately 90% of a levodopa dose is converted by COMT to 3-O-methyl-levodopa (3-OMD) which competes with levodopa at the level of the blood-brain barrier for transport. Thus, an additional strategy to further inhibit peripheral levodopa metabolism and increase the delivery of levodopa to the brain is the administration of a COMT inhibitor [4, 18-20]. The two inhibitors used for this purpose are tolcapone and entacapone [21]. Actually, it is marketed the opicapone [19].
1.2 Properties of COMT isoforms
Genetic and pharmacological manipulation of COMT activity has demonstrated positive effects in human studies [3]. These studies provide validation for COMT inhibition as a promising avenue for treatment of cognitive deficits in schizophrenia and PD, although no distinctions have yet been made with regards to selective MBCOMT or SCOMT inhibition [21, 22]. As referred above, MBCOMT has much lower capacity and Km value than the soluble form but a
higher affinity to catecholamines [6, 13]. Thus, at high substrate concentrations, the SCOMT activity increases. On the other hand, when concentrations of substrate are low, MBCOMT is the predominant isoform. Furthermore, there are other differences between both isoforms, such as isoelectric point (pI) values, which is 5.2 for SCOMT and 6.2 for MBCOMT [11].
1.2.1 Biosynthesis of SCOMT and MBCOMT
In last two decades, the yeast Pichia pastoris (P. pastoris) has been used frequently a expression system for recombinant protein production [6, 10]. P. pastoris is a single-cell microorganism and by this way, it is easily manipulated and cultivated [23, 26]. The advantages of this system include growth up to high cell densities quantity on defined minimal medium, high expression level of heterologous proteins and efficient secretion of extracellular proteins. Among P. pastoris, more remarkable features are the promoter derived from the alcohol oxidase I (AOX 1). This yeast has two alcohol oxidase genes, AOX 1 and AOX 2 of which AOX 1 is much more strongly transcribed than AOX 2, making it a great advantage when induction is effected by methanol. [6, 26, 33].
For expression of the soluble COMT (SCOMT), different expression systems have been used such as transfected mammalian cells [16], insect cells [16] (via mammalian and baculovirus vectors), plant cells (via a potyvirus) [23] and prokaryotic cells, such as Escherichia coli (E.
coli) [25] that is a Gram-negative bacterium. Usually the major biorecombinant resource of
SCOMT, for specific biopharmaceutical and neurological trials, is E. coli such as E. coli SG 13009 and BL21 [7, 24, 25, 33]. It should be noted that the optimization of environmental conditions such as temperature, pH, inducer concentration and stabilizers concentration are essential for the production of recombinant proteins to ensure a good quality of the target product [24].
In relation to MBCOMT recombinant protein, several expression systems have been explored to produce high amounts of this enzyme. It´s expression was successfully reached using prokaryotic hosts such as E. coli BL21 and SG 13009 strains [6, 33]. Another system used for expression of recombinant protein is Brevibacillus choshinensis that is a gram-positive microorganism. This system is well suited for secretory production of heterologous proteins with high efficiency as it produces a small amount of extracellular proteases [63]. Beyond aforementioned systems there are others that are also used for recombinant expression such as, Sf9 insect cells, transfected human embryonic kidney fibroblast cell lines, human HeLa, and hamster BHK cells. However, in some systems is impossible to express this recombinant protein, since its hydrophobic sequence can be toxic to host [6]. Another system used of MBCOMT recombinant protein is P. pastoris. This recombinant production, P. pastoris has been described as an attractive host for the production of correctly folded and inserted membrane proteins [6].
1.3 Chromatography
Chromatography is a method mainly used in separating components of a sample, whose aim is not only removal of unwanted contaminants, but also the concentration of the desired protein and the transfer to an environment where it is stable and in a form ready for the intended application [34].
Nowadays, chromatographic processes can be differentiated in two types, the analytical and preparative chromatography. Regarding analytical chromatography the principal aim is the rapid detection of specific components through the direct signal acquisition in order to calculate its concentration by a calibration curve [35]. On the other hand, in preparative chromatography it is required the injection of large quantities of sample in the column in order to obtain a given amount of pure product with high recuperation yield [35].
Currently, many chromatographic techniques are available, such as ion exchange chromatography (IEC), hydrophobic interaction chromatography (HIC), affinity chromatography (AC), reverse phase chromatography (RPC) and gel filtration chromatography (GFC). These different types of chromatography are studied according to different types of interactions involved between solutes in the mobile phase and the stationary phase [8, 28, 36-38]. Therefore, an insoluble matrix is used as stationary phase and is packed into a column and the mobile phase is pumped through the system [36, 39]. Column chromatography is the most common physical configuration, in which the stationary phase is packed into a tube, a column, through which the mobile phase, the eluent, is pumped, such as demonstrated in Figure VI [34, 35].
Generally, a typical chromatographic procedure presents five major stages, equilibrium phase, sample injection, washing of non-retained species, elution of molecules adsorbed in matrix and regeneration (Figure VII) [39]. Briefly, the equilibrium phase aims to maintain optimal conditions at the mobile phase that allow the binding of the target biomolecule to the stationary phase [39]. The sample injection consists in the insertion of a certain quantity of a complex mixture, which contains the target protein, into a stationary phase [38, 39]. The sample is transported by mobile phase and will be distributed between the mobile phase and stationary phase according to its affinity. In the washing stage, impurities that not interact within stationary phase are removed from the column using the same buffer of the column equilibrium [36, 38]. The elution of the retained species is achieved using a buffer that decline the strength of interactions established between the matrix and the target biomolecule. Thus, the proteins strongly adsorbed move more slowly through the column than the weakly bound biomolecules [38]. Lastly, the regeneration step is a very important cleaning process that maintains the binding capacity, selectivity and lifetime of the chromatographic support. The cleaning procedure depends on matrix type but in general are used highly acid or basic solutions, low or high-salt concentrated solutions or organic solvents [38].
Figure VII - Representation of the phases of chromatographic procedure (adapted from [39]).
1.3.1 Chromatographic Matrix
A chromatographic matrix should be insoluble in the buffer, hydrophilic, easily activated and coupled to a ligand in order to explore different interactions, depending of the ligand nature. It should also have large pores accessible to the protein, have a large surface area to increase
the binding capacity, and be physically and chemically stable to withstand the conditions during derivatization and sterilization [39].
A variety of materials have been used as matrices. These include inorganic materials such as glass, silica, and hydroxyapatite [40]; synthetic organic polymers such as polyacrylamide, polystyrene and polysaccharides [38]. The most commonly used supports are matrices derived from synthetic organic polymers with based agarose, such a Sepharose (Figure VIII) [38, 39]. It should be noted that the agarose is widely used for adsorption chromatography because of its reasonable rigidity, stability, and the ease of surface modification to couple functional groups [39, 40]. In addition, an advantage to use agarose or monoliths is that these supports are fairly easy to prepare in a variety of shapes and sizes [40].
Figure VIII - Structure of Sepharose (adapted from [38]).
1.3.2 Chromatographic methods for SCOMT and MBCOMT purification
The development of techniques and methods for the separation and purification of proteins has been essential for many of the recent advancements in biotechnology research. The choice of a suitable purification technique depends on location, characteristics and desired purity of the target protein [34]. The purification procedure presents high efficiency how lower the number of steps. Thus, it is possible to obtain high yields and the suitable quality and purity [34, 35].
Nowadays, various chromatographic procedures can be applied in purification of proteins. Thus, the chromatography has become one of the preferential techniques for the SCOMT purification due to its high resolving power [16, 42, 43, 45].
1.3.2.1 Hydrophobic interaction chromatography
The HIC is a powerful separation technique where protein purification is based in hydrophobic interactions between hydrophobic ligands immobilized on matrix and non-polar regions on proteins surface. This technique is generally apply immediately after a salt precipitation step [41 - 43]. The adsorption increases with high salt concentration in the mobile phase and the elution is achieved by decreasing the salt concentration of the eluent [37, 41, 42]. The main factors affecting protein chromatographic behavior in HIC are protein hydrophobicity, their surface hydrophobicity distribution and molecular size [41, 43].
This purification process has been extensively applied in purification of SCOMT. This isoform was totally retained on several hydrophobic matrixes and by decreasing the ammonium sulphate concentration, the SCOMT was isolated with a basal loss of specific activity [42]. For instance, butyl-Sepharose resin with an optimized elution gradient revealed the best performance relative to the purity ratio. In addition, the less hydrophobic adsorbent (epoxy-Sepharose) is considered as a last resort due to the high ammonium sulphate concentration needed, which will compromise the protein activity [42].
Regarding to MBCOMT, the purification process is more difficult than SCOMT, because it is highly desirable transfer the protein for a more hydrophobic environment when it is working with a membrane protein. This can be achieved by treating the sample with detergents [5, 13, 37]. Detergents are amphipathic molecules, consisting of a polar head group and a hydrophobic chain that solubilizes membrane proteins by creating a mimic of the natural lipid bilayer environment. For this purpose, the type of detergent can be ionic, non-ionic, zwiterionic or bile acid salts [13]. In this way, it is essential to adjust the detergent type and concentration to the characteristics of target biomolecule in order to avoid irreversible structural loss [37]. Another study to purify the MBCOMT from crude Brevibacillus
choshinensis cell lysates was conducted by comparing different hydrophobic ligands such as
octyl, butyl and epoxy [13, 37]. In case of octyl and butyl ligands, the MBCOMT adsorption was performed at moderate salt concentrations and the elution was promoted by using 1 % Triton X-100. On the other hand, higher salt concentrations were used for protein adsorption in the case of epoxy ligand and its elution was promoted also by using 0.8 % Triton X-100 [37].
1.3.2.2 Ion exchange chromatography
The IEC is one technique of the most chromatographic methods applied in the purification of soluble proteins and membrane protein [49]. The IEC allows the separation of biomolecules with high degree of resolution according to differences in their surface charge at specific pH value [38, 49]. The main advantages of this technique is the capacity to purify biomolecules with positive or negative charge [39, 49].
One of chromatographic techniques that have gained great importance in isolating or purifying the SCOMT was IEC [11]. This chromatography process seeking is not only removal of unwanted contaminants, but to promote the concentration of the desired protein [30]. Actually, there are already some purification processes for MBCOMT using Resource Q column as chromatographic support. This strategy involved the MBCOMT solubilisation with Triton X-100 and its purification was also performed using Triton X-X-100 [5]. So, MBCOMT adsorption can be promoted with application of 0.5 % Triton X-100 in 10 mM Tris HCl. The elution of isoform was performed by an increase in ionic strength [5, 11].
1.3.2.3 Affinity chromatography
The AC is a high-resolution technique that separating proteins based in highly specific biological interactions between the protein and an affinity ligand, providing elevated selectivity [16, 39]. These methods require often the addition of an affinity tag to protein during vector construction step, which facilitates target protein binding to chromatographic matrix [16]. Consequently, affinity tag removal is necessary after the purification step, which usually implies a significant reduction in process yield and irreversible activity losses [8]. This strategy was successfully described for the isolation of biologically active SCOMT. The elution was performed an increasing NaCl stepwise gradient.
1.3.2.4 Immobilized metal-affinity chromatography
Immobilized metal-affinity chromatography (IMAC) is an affinity technique of chromatographic separation based in affinity between the immobilized metal ions on a solid matrix and the biomolecule in solution [33, 44, 45]. This affinity results of reversible linkages formed between metal ion (the most frequently used are Cu (II), Ni (II), Zn (II), Co (II) and Fe (III)) and electron donor groups located on the surface of the proteins, mainly histidine residues, often introduced into a target protein as a N- or C- terminal peptide ‘tag’ [33, 46, 47]. In general, the biomolecules are retained in IMAC using equilibrium buffer without imidazole or at low concentrations between 1 to 10 mM and the elution is usually achieved by increasing the imidazole concentration [33]. Usually the buffers containing NaCl in order to reduce nonspecific electrostatic interactions. Generally, in IMAC, through the competition with nickel ions, the imidazole is responsible for eluting the proteins and when present at low concentrations in the binding buffer, it may prevent the binding of host proteins with exposed histidines, allowing the removal of contaminants in the flowthrough during the injection of the sample [48]. This methodology is extremely efficient and selective for the direct capture of hexahistidine tagged SCOMT from recombinant P. pastoris lysates [33]. The adsorption was
achieved with increase stepwise concentration of imidazole (300 mM imidazole). The best strategy allowed recovering SCOMT at 300 mM imidazole in a highly purified fraction with a purification fold of 81 and a bioactivity recovery of 57.35% [33].
1.3.3 Ionic Exchange Chromatography
IEC is one of the most frequently used techniques for purification of proteins, peptides, nucleic acids and other charged biomolecules, since it is obtained high resolution and separations with high loading capacity [11, 38, 34, 39] .
In general, in IEC, the charged sample is first loaded into the column at low ionic strength interacting with oppositely charged ligands [32, 38]. After, low affinity proteins are quickly eluted to the column, while the high affinity proteins are retained for further elution. The elution is usually performed by increasing the salt concentration or suitable modification of pH [31, 32]. For this purpose the increase of ionic strength in the eluent affects the retention, since the high salt concentration favour the competition by the solutes, resulting in the elution of the components of lower affinity. Finally, the high ionic strength wash removes any ionically bound proteins before re-equilibration (Figure IX) [32, 38, 39].
Figure IX – A typical profile from ion exchange chromatography (adapted from [32]).
Whereas, the pH of the elution buffer determines the molecule ionization state. A protein that has no net charge at a pH equivalent to its isoelectric point (pI) will not interact with a charged medium. However, at a pH above its isoelectric point, the protein is negatively
(AEC). On the other hand, at pH below its pI, the protein is positively charged, and will bind to a negatively charged medium by cation exchange chromatography (CEC) (Figure X) [32, 38, 39].
Figure X - Effect of pH on protein binding and elution patterns in Ionic Exchange Chromatography
(adapted from [32]).
This chromatographic procedure has revealed to be quite efficient to separate both soluble proteins and membrane proteins (MPs) due to numerous advantages such as purifications in large scale, relatively low cost and use of any neutral detergents, which reduces the risk of MPs instability during the chromatographic step [31, 32, 39].
Overall, IEC requires a stationary phase, usually composed by hydrated insoluble polymers such as cellulose or Sephadex, to which is coupled an ion exchanger group, that can be cationic or anionic. Thus, this type of chromatography can be classified into CEC or CEA [38]. For instance, in AEC, the stationary phase carries positively charged functional groups that are capable of binding anions (e.g. ionized carboxylic acids). The mobile phase usually contains a buffer to maintain stable pH and varying the salt concentration to control the retention of sample ions (counter ions) [38]. In relation to these ion exchangers can be classified as strong and weak exchangers based on the difference between the functional groups (Table I). The ion exchangers are strong due the fact that they are completely ionized at a specific pH, while the weak exchangers have a degree of ionization dependent of pH. An example of a weak anion exchanger is usually diethylaminoethyl (DEAE) [32, 35, 38].
Table I - Types of ion exchangers (adapted from [32]).
1.3.4 Monolithic supports
Currently, the chromatographic technology is a widely used method in the purification of biomolecules, where one of its main objectives is to develop rapid and efficient separations with high binding capacity, as well as, apply specific ligands to improve the selectivity for the target molecule [50, 51, 53]. An example of this technology is the monolithic chromatography. Monoliths are a special type of chromatographic column, considered the material of choice for the purification and analysis of proteins, plasmid DNA and viruses [51, 52, 53, 54, 56, 58].
The monolithic supports have been increasingly used due to its advantages over traditional matrices, such as, high porosity, high binding capacity for extremely large molecules and convective mass transport [51, 52, 54, 59]. The fact that the monoliths present channels with high porosity makes the binding capacity increased. In fact, pore dimension is correlated with the exclusion limit, which defines the size range of molecules that can enter or be excluded from the pore. In reality, the pore size of a matrix is inversely correlated to its surface area, which in turn directly affects the amount of immobilized ligand [40, 50, 51, 58]. Therefore, another advantage of monolithic columns is low absolute surface area, but a high adsorption due to their channel structure, allowing a high dynamic binding capacity [40, 51]. The separation of biomolecules is another advantage and dominantly happens by convective mass transport that allows high flow velocity, in this way, all the mobile phase is forced to flowthrough to the channels via convection, causing high throughput purifications [40, 50, 57, 58]. Lastly, the chromatographic columns need to be short, supporting higher flow rates, without sacrificing resolution or the bioactivity of the target molecule [40, 52].
1.3.4.1 CarbonylDilmidazole Monolith
The CarbonylDilmidazole (CDI) monolithic support allows the convective mass transport of the macromolecules, offering a great binding capacity due to their single structure with a highly interconnected network of a large diameter channels [50]. This support, when activated, can be used in ligand immobilization by means of a nucleophilic substitution, resulting in a stable amide linkage, as seen in Figure XI [57].
Figure XI - Immobilization of ligands by CarbonylDilmidazole monolithic support (adapted from [57]).
However, the CDI disc without modification has been successfully used in the separation of plasmid DNA (pDNA) isoforms due to the imidazole functional groups by using a decreasing stepwise ammonium sulphate ((NH4)2SO4)gradient [61]. Should be pointed out that CDI ligands
can establish several interactions accountable for the biorecognition of the biologically active pDNA isoform, namely hydrophobic interactions, van der Waals forces and hydrogen bonds, allowing the elimination of host impurities present in the lysate sample [52, 61]. In fact, it is important to refer that the chemical composition of chromatographic supports determines the interactions established with the target molecule, allowing its retention whereas undesirable molecules are eluted [50].
1.3.4.2 Histamine Monolith
Histamine monolithic support consists in a CDI monolith modified with the histamine ligand. The histamine ligand is derived from the decarboxylation of L-histidine amino acid, containing
Figure XII - Schematic representation of immobilized histamine ligands (adapted from [56]).
This monolithic support has been successfully used also in the pDNA purification due to physicochemical properties and versatility of the ligand [55, 56, 59]. For instance, the Histamine monolith showed to be a multifaceted column to purify the sc pDNA from a lysate sample by different strategies (the simple purification strategy with ascending sodium chloride gradient and the combined purification strategy with ascending sodium chloride and then descending ammonium sulphate gradient) [56].
Although the histamine ligand has not yet been tested in the purification of proteins, the L
-histidine amino acid has been successfully used as a selective and efficient ligand for purification of various proteins and peptides [40]. Properties such as mild hydrophobicity, weak charge transfer, asymmetric carbon atom, and wide pKa values are characteristics that
make histidine a potential ligand for protein purification [40, 56].
1.3.4.3 Agmatine Monolith
Agmatine monolithic support was also prepared from a CDI monolith modified with the agmatine ligand. Agmatine ligand is derived from the decarboxylation of arginine amino acid, containing basic guanidinium group, as seen in Figure XIII [56, 58, 62].
This monolith has been used in microRNAs purification processes by exploiting the versatility of this ligand through three different binding and elution strategies based on increased NaCl and decreased (NH4)2SO4 stepwise gradients, in order to obtain the final product with high
purity degree [58]. It was also used in the pDNA purification, working under two elution strategies, by descending (NH4)2SO4 gradient and by ascending NaCl gradient, combining ionic
Justification and Objectives
The main objective of this work is to explore the chromatographic conditions more appropriate for the recognition of SCOMT_6His protein by manipulating the elution buffer composition (type and salt concentration), using monolithic supports for the first time. To reach this propose, two purification strategies were developed.
Firstly, the comparison and assessment of the chromatographic behaviour of SCOMT_6His using Q-Sepharose as anion exchanger was performed in order to analyse the Q-Sepharose performance in terms of binding and elution conditions for SCOMT_6His recovery and pre-purification. Thereafter, the behavior of the pre-purified sample was analysed in three monolithic columns (CDI, Histamine and Agmatine) to explore different elution strategies by increasing and decreasing of sodium chloride and ammonium sulphate concentrations and increase the final purity degree of the SCOMT_6His protein.
After choosing the most promising monolith, the lysate sample was filtered and directly injected in the monolith, in order to obtain a greater recovery of the target protein with satisfactory purity degree in comparison to the other strategy.
Chapter II
Materials and Methods
2.1 Materials
Ultrapure reagent-grade water for ÄKTA™ avant was obtained with a Mili-Q system (Milipore/Waters). The easy select expression kit for expression of recombinant proteins using pPICZα vector in P. pastoris and zeocin (200 μL) were obtained from Invitrogen (Carlsbad, CA). Yeast extract, glucose, agar, tryptone, sorbitol, yeast nitrogen base (YNB), biotin, methanol, Cysteine (L-), bovine serum albumin (BSA), S-(5’-adenosyl)-L-methionine chloride
(SAM) and epinephrine (bitartrate salt) were obtained from Sigma Sigma-Aldrich (St. Louis, MO). Glycerol and sodium chloride (NaCl) was obtained from Himedia (Mumbai, India) and from Panreac (Barcelona, Spain), respectively. NZYcolour Protein Marker II used for estimation of subunit molecular weight was purchased in NZYTech (Lisboa, Portugal). Anti-rabbit IgG alkaline phosphatase secondary antibody was purchased on GE Healthcare Biosciences (Uppsalla, Sweden). Monoclonal rabbit anti-COMT antibody was produced in BIAL (S. Mamede do Coronado, Portugal). Acrylamide 30%/Bis solution was obtained from BioRad (Hercules, CA). Tris(hydroxymethyl)aminomethane (Tris) and CAPS were obtained from Fisher Scientific (Epson, United Kingdom). All other chemicals were of analytical grade and used without further purification. The agmatine monolith, histamine monolith and CDI monolith were kindly prepared and provided by BIA Separations (Ajdovščina, Slovenia).
2.2 Plasmids, bacterial strains and media
Briefly, the DNA fragment coding for SCOMT was obtained from the pET101/D-hSCOMT plasmid by polymerase chain reaction (PCR) using specific primers (forward primer, 5’ AAC TCG AGA AAA GAA TGG GTG ACA CCA AGG AGC AG 3’ and reverse primer, 5’ AAC TCG AGT CAG TGA TGG TGA TGG TGA TGG GGC CCT GCT TCG CTG CCT G 3’) for cloning in which the reverse primer was designed in order to introduce a hexahistidine tag in SCOMT carboxyl-terminal. The PCR conditions were as follows: denaturation at 95 ºC for 5 min, followed by 30 cycles at 95 ºC for 30 s, 60 ºC for 30 s and 72 ºC for 1 min, and a final elongation step at 72 ºC for 5 min. The amplified DNA was purified by low melting agarose gel electrophoresis, digested with Sac I and cloned into the vector pPICZ𝛼 (previously digested with Sac I) by T4
in plates with Low-salt Luria-Bertani containing 25 μg/mL Zeocin and colonies were screened for the presence of the construct pICZ𝛼A-hSCOMT_His6. Therefore, some colonies were inoculated in 2.0 mL of Low-salt Luria-Bertani and grown at 37 ºC and 250 rpm overnight. From these cultures, highly purified plasmids were prepared using NzyMiniprep (Nzytech, Lisboa, Portugal) and were then subjected to DNA sequence analysis to confirm the identity of the amplicon, orientation and frame. Since the sequence was confirmed to correspond to human SCOMT gene with the six histidines addition in its carboxyl-terminal, the cloned plasmid was digested with Sac I and introduced into freshly made P. pastoris X-33 competent cells by electroporation at 2.5 Kv (2500 V), 25 µF and 1000 Ω [11, 33]. After confirming that the X-33 integrant presented a methanol utilization phenotype plus (Mut+), the stable
occurrence of the expression cassette was verified in the colonies genomic DNA by PCR. This process was carried out according to manufacturer’s instructions and as previously described [11, 33].
2.3 Recombinant SCOMT_6His production and recuperation
Recombinant SCOMT_6His production was performed using P. pastoris X-33 cells containing the expression construct pICZαA-hSCOMT_His6 according to the following protocol: cells containing the expression construct were grown for 72 hours at 30 ºC in YPD plates containing 200 μg/mL Zeocin. A single colony was inoculated in 100 mL of BMGH medium (100 mM potassium phosphate buffer, pH 6.0, 1.34 % YNB, 4 × 10-5 % biotin and 1 % glycerol) in 500 mL
shake flasks and grown overnight at 30 ºC and 250 rpm to a cell density at 600 nm (OD600) of
6. Posteriorly, since the inoculation volume was fixed to achieve an initial OD600 of 1, an
aliquot of the fermentation in the BMHH medium (125 mL) (100 mM potassium phosphate buffer, pH 6.0, 1.34 % yeast nitrogen base (YNB), 4 × 10-5 % biotin and 0.5 % methanol) was
collected and centrifuged (500 x g, 5 min at room temperature). After centrifuging the cells and to ensure that all glycerol was removed, the cells were ressuspended in the induction medium and added to 500 mL shake-flasks to a total volume of 125 mL. The fermentations were carried out during 24 hours at 30 ºC and 250 rpm and were supplemented with methanol 1%. Then, the cells were harvested by centrifugation (1500 x g, 10 min, 4 ºC) and stored at – 20 ºC until use. Thereafter, cells were lysed in equilibrium buffer (150 mM NaCl, 10 mM DTT, 50 mM Tris, 1 mM MgCl2, pH 8.0) at a ratio of 1:2:2 (1 g cells, 2 mL lysis buffer and 2 g glass
beads). Lysis was accomplished through the application of a sequential procedure with glass beads of 7 cycles of vortexing for 1 min with 1 min of interval on ice. Subsequently, the mixture was centrifuged (500 x g, 5 min, 4 ºC) and the pellet obtained was ressuspended in the chromatographic binding buffer (500 mM NaCl, 50 mM Tris and 1 mM MgCl2 at pH 7.8)
2.4 Pre-purification by Anion Exchange Chromatography
Chromatographic assays were performed at temperature 6 ºC in ÄKTA Avant system with UNICORN 6 Software (GE Healthcare, Uppsala, Sweden) equipped with a 2 mL injection loop. All buffers pumped into the system were prepared with Mili-Q system water, filtered through a 0.20 μm pore size membrane (Schleicher Schuell, Dassel, Germany) and degassed ultrasonically.
Q-Sepharose (GE Healthcare Biosciences) was packed according to company guide-lines (20 mL of gel volume) into a C 16/20 [16 mm (diameter) x 200 mm (length)] glass column purchased from GE Healthcare Biosciences. Screening experiments were performed at different salt concentrations in order to assess the ideal sodium chloride concentration required for SCOMT_6His retention. The column was initially equilibrated with 10 mM Tris-HCl buffer pH 7.8. Aliquots of recombinant SCOMT_6His containing supernatant (3 mL) were injected into column at 1 mL/min with 10 mM Tris-HCl pH 7.8. The elution of unretained species occurred with an increasing sodium chloride gradient from 0 mM to 100 mM (3 CV). After elution, sodium chloride concentration was gradually increased from 100 mM to 310 mM (3.5 CV) to ensure that the protein was completely bound to the column. Subsequently, sodium chloride concentration in mobile phase was increased to 450 mM in a step mode (2.5 CV). Finally, a washing step was applied with 1 M of sodium chloride (2 CV) in a step mode. In all chromatographic runs, conductivity was continuously monitored, as well as absorbance at 280 nm. Fraction volumes of 3 mL were collected and pooled according to the obtained chromatographic profile. Finally, samples were concentrated and desalted with 1 mL 10 mM Tris-HCl pH 7.8 with Vivaspin concentrators (10.000 MWCO) and conserved at 4 ºC until further analysis.
2.5 Purification by Monoliths
All chromatographic experiments were carried out in an ÄKTA Avant system accomplish with UNICORN 6 Software (GE Healthcare, Uppsala, Sweden). All buffers were filtered through a 0.20 μm pore size membrane (Schleicher Schuell, Dassel, Germany), and degassed ultrasonically. The system was run at a flow rate of 1 mL/min, and monitored at 280 nm. Initially, the CDI monolith was equilibrated with 3M (NH4)2SO4. The sample clarified by
Q-Sepharose was applied onto the column using a 200 μL loop at a flow rate of 1 mL/min. After the elution of unbound species, the ionic strength of the buffer was decreased to 0 M of (NH4)2SO4 in 10 mM Tris-HCl buffer (pH 7.8). After that, the assay under ionic condition was
also performed. The CDI column was equilibrated with 10 mM Tris-HCl buffer (pH 7.8) and after the sample injection the ionic strength was increased to 1.5 M of NaCl in 10 mM Tris-HCl
repeated for the Histamine monolith. The Agmatine monolith was used only in ionic elution conditions. The Agmatine column was equilibrated with 10 mM Tris-HCl buffer (pH 7.8) and after the sample injection the sodium chloride concentration in mobile phase was increased to 1.5 M in a step mode. Finally, a washing step was applied with 3 M of NaCl in 10 mM Tris-HCl buffer (pH 7.8) in a step mode.
Finally, samples were concentrated and desalted with 3 mL 10 mM Tris-HCl applying Vivaspin concentrators (10.000 MWCO) and conserved at 4 ºC until further analysis.
2.6 Total protein quantification
The protein content in lysate and purified samples was measured by the Pierce BCA Protein Assay Kit (Thermo Scientific, USA) on a 96 well plate, and a specific volume of working reagent (WR) was prepared from the kit according to the number of existing samples. It was used BSA as the standard and calibration control samples (5–2000 µg/mL).
2.7 SDS-PAGE and Western blot
Reducing Sodium Dodecyl Sulphate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Western blot trials were performed, respectively according to the method of Laemmli [64] and the literature described above [42]. Samples (30 µL) to analyse in SDS-PAGE were treated by adding 10 μL of a reduction buffer. Samples were boiled in a loading buffer (500 mM Tris-HCl (pH 6.8), 10% (w/v) SDS, 0.02% (w/v) bromophenol blue, 0.2% (v/v) glycerol, and 0.02% (v/v) β- mercaptoethanol) and were denatured at 100 ºC for 5 min. The run on 4% stacking and 12.5% resolving gels containing 0.1% SDS, with a running buffer (25 mM Tris, 192 mM glycine, 0.1% (w/v) SDS) at 120 V for 1h 40 min. After electrophoresis, one gel was stained by Comassie brilliant blue and the other gel was transferred to a polyvinylidene difluoride (PVDF) membrane, in order to perform the western blot experiments.
For Western blot, the proteins were transferred over a 30 min period at 750 mA at 4 ºC in a buffer containing 10 mM CAPS. After the blotting, the membranes were blocked with TBS-T (pH 7.4) containing 5% (w/v) milk for 60 min at room temperature, washed 3 times during 15 minutes and exposed overnight at 4 ºC to a rabbit anti-rat SCOMT polyclonal antibody, that cross reacts with the human protein, at 1:2000 dilution in TBS-T 1%. The membranes were washed three times (15 min each) with TBS-T and adherent antibody was detected by incubation for 1 h with an anti-rabbit IgG alkaline phosphatase secondary antibody at 1:40000 dilution in TBS-T 1%. The PVDF membranes were air dried, incubating with 1 mL of ECF for 5 min and enhanced by exposure to chemiluminescence’s detection.
2.8 SCOMT_6His enzymatic assay
The methylation efficiency of SCOMT_6His was evaluated by measuring the amount of metanephrine formed from epinephrine. After processing the samples, the metanephrine levels in the different samples obtained in the Q-Sepharose assays were determined using a HPLC with coulometric detection [65].
Briefly, the chromatographic analysis was performed using a HPLC model Agilent 1260 system (Agilent, Santa Clara, USA) equipped with an auto sampler and quaternary pump coupled to an ESA Coulchem III detector (Milford, MA, USA). Chromatographic separation was achieved on an analytical column Zorbax 300SB C18 RP analytical column (250 × 4.6 mm with i.d. of 5 μm) (Agilent, Santa Clara, California, USA). The chromatographic method was developed using as mobile phase (0.1 M NaH2PO4, 0.024 M citric acid monohydrate, 0.5 mM sodium octyl sulpahte
and 9% (v/v) acetonitrile) and the column effluent was monitored with an electrochemical detector in a coulometric mode, which was equipped with high sensitivity dual electrode analytical cell (electrodes I and II) using a procedure of oxidation/reduction (analytical cell #1: +410 mV; analytical cell #2: −350 mV). The method sensitivity was set at 1 μA, the flow-rate applied was 1 mL/min and the column temperature was optimized to 30 ºC. The chromatograms were obtained by monitoring the reduction signal of the working electrode where metanephrine retention time was around 8 min. Finally, the metanephrine content in samples was measured using metanephrine standards as a calibration control. The calibration curve was used y= 3521,2871x + 367,0215 [65].
Chapter III
Results and Discussion
In recent years, several attempts have been performed to obtain a large quantity of enzymatically active and pure SCOMT protein. Therefore, it was important to develop an appropriate strategy to reach this objective, ie, to develop purification strategies that allow to recover high levels of pure recombinant enzymatic product in an active form [5, 7]. According to the literature, the SCOMT protein has been subjected to numerous purification procedures. In fact, SCOMT purification by AEC combined with others chromatographic steps led to successful resolving of its atomic structure [11, 16, 28, 31].
Therefore, this work intends to establish two chromatographic strategies to purify the SCOMT_6His protein through the use of Q-Sepharose column in order to clarify the cell lysates of P. pastoris and subsequently evaluate the applicability of affinity chromatography by the use of three monolithic supports (CDI, Histamine and Agmatine).
3.1 Production of SCOMT_6His
SCOMT_6His biosynthesis from P. pastoris X-33 cells, containing the expression construct pICZα A-hSCOMT_His6, was previously optimized in order to enhance COMT activity recovery [33]. The SCOMT_6His production was monitored by measuring cell density at 600 nm (OD600)
over time. Its maximum output was reached after 24 hours as seen in Figure XIV.
![Figure I - A typical reaction catalysed by COMT. SAM: S-adenosyl- L -methionine; SAH: S-adenosyl- L - -homocysteine; Mg 2+ : Magnesium (adapted from [4])](https://thumb-eu.123doks.com/thumbv2/123dok_br/18180251.874444/21.892.239.624.717.945/figure-reaction-catalysed-adenosyl-methionine-adenosyl-homocysteine-magnesium.webp)
![Figure III - Representation of some structures of first generation COMT inhibitors (adapted from [3])](https://thumb-eu.123doks.com/thumbv2/123dok_br/18180251.874444/25.892.181.706.481.789/figure-iii-representation-structures-generation-comt-inhibitors-adapted.webp)
![Figure V - Representation of metabolic pathway of L-Dopa (adapted from [1]). L-Dopa: Levodopa; DDC:](https://thumb-eu.123doks.com/thumbv2/123dok_br/18180251.874444/26.892.171.642.671.927/figure-representation-metabolic-pathway-dopa-adapted-dopa-levodopa.webp)
![Figure VI - Representation of a typical column apply in chromatography (adapted from [38])](https://thumb-eu.123doks.com/thumbv2/123dok_br/18180251.874444/29.892.171.726.759.1063/figure-vi-representation-typical-column-apply-chromatography-adapted.webp)
![Figure VII - Representation of the phases of chromatographic procedure (adapted from [39])](https://thumb-eu.123doks.com/thumbv2/123dok_br/18180251.874444/30.892.145.684.581.889/figure-vii-representation-phases-chromatographic-procedure-adapted.webp)
![Figure IX – A typical profile from ion exchange chromatography (adapted from [32]).](https://thumb-eu.123doks.com/thumbv2/123dok_br/18180251.874444/34.892.176.696.662.979/figure-ix-typical-profile-ion-exchange-chromatography-adapted.webp)
![Figure X - Effect of pH on protein binding and elution patterns in Ionic Exchange Chromatography (adapted from [32])](https://thumb-eu.123doks.com/thumbv2/123dok_br/18180251.874444/35.892.329.607.204.576/figure-effect-protein-binding-elution-patterns-exchange-chromatography.webp)
![Table I - Types of ion exchangers (adapted from [32]).](https://thumb-eu.123doks.com/thumbv2/123dok_br/18180251.874444/36.892.102.753.141.356/table-i-types-ion-exchangers-adapted.webp)
![Figure XI - Immobilization of ligands by CarbonylDilmidazole monolithic support (adapted from [57])](https://thumb-eu.123doks.com/thumbv2/123dok_br/18180251.874444/37.892.184.728.331.545/figure-xi-immobilization-ligands-carbonyldilmidazole-monolithic-support-adapted.webp)
