DANIELA BACCELLI SILVEIRA MENDONÇA
SUPRESSÃO DA RESPOSTA DO HIF-1
PELO NF-
B VIA COMPETIÇÃO PELA
LIGAÇÃO COM O COATIVADOR TRANSCRICIONAL p300
Tese apresentada ao Programa de
Pós-graduação
Strictu
Sensu
em
Ciências
Genômicas e Biotecnologia da Universidade
Católica de Brasília como requisito para
obtenção do Título de Doutor em Ciências
Genômicas e Biotecnologia.
Orientador:
Prof. Dr. Francisco José Lima
Aragão.
Co-orientador:
Prof. Dr. Lyndon Frederick
Cooper.
À Deus pela vida e por todas as
oportunidades a mim oferecidas.
Ao querido Gustavo pelo carinho, amor,
compreensão, paciência, e por todo auxílio
que foram essenciais para que eu pudesse
terminar mais essa etapa.
Aos meus queridos pais Mac Tulio e Maria
Inez, exemplos de força e determinação,
agradeço pela educação, amor e apoio
incondicionais.
AGRADECIMENTO ESPECIAL
Aos meus Orientadores, Prof. Dr. Francisco José Lima Aragão e Prof. Dr. Lyndon
Frederick Cooper, agradeço pela confiança em mim depositada, pela disponibilidade
de tempo, pelos conhecimentos compartilhados e por terem disponibilizado os
recursos necessários para o término desse projeto
AGRADECIMENTOS
À Universidade Católica de Brasília.
À Embrapa Recursos Genéticos e Biotecnologia.
À Universidade da Carolina do Norte em Chapel Hill-EUA.
À Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – CAPES – pela
bolsa de doutorado Sandwich que me permitiu expandir meus conhecimentos e na
Universidade da Carolina do Norte em Chapel Hill-EUA.
À todos os professores do Programa de Pós-graduação em Ciências Genômicas e
Biotecnologia da Universidade Católica de Brasília.
Aos funcionários do Programa de Pós-graduação em Ciências Genômicas e
Biotecnologia da Universidade Católica de Brasília, especiamente ao Francisco
Fábio Gomes da Costa.
Aos amigos de pós-graduação pelos momentos que passamos juntos e aprendemos
uns com os outros.
À Sara Valencia, reponsável pelo andamento de todas as atividades no Laboratório
de Biologia Óssea e Mineralização da Universidade da Carolina do Norte, e que
muito ajudou para que nossos trabalhos no laboratório funcionassem perfeitamente.
À Gidget Jenkins e Martha Taylor do departamento de Prótese da Faculdade de
Odontologia da Universidade da Carolina do Norte, por também nos ajudarem em
tudo que fosse necessário durante nossa estadia nos Estados Unidos.
Aos amigos da Universidade da Carolina do Norte, Gustavo e Greice, Ricardo e
Patrícia, Luiz e Silvana, Sodsi “Nid” Wirojchanasak e Ghadeer Thalji.
RESUMO
MENDONÇA, Daniela Baccelli Silveira. Supressão da resposta do HIF-1
pelo
B via competição pela ligação com o coativador transcricional p300. 2010.
138p. Tese. Ciências Genômicas e Biotecnologia – Universidade Católica de
Brasília, Brasília, 2010
dependente. Conclui-se que a sinalização inflamatória mediada pelo NF-
B é capaz
de bloquear a transativação do HIF-1
nos sítios HRE dos genes que respondem ao
HIF-1
através de uma competição direta pela ligação ao p300. A inflamação pode
influenciar o nicho de células-tronco e a regeneração tecidual por interferir com a
resposta das células à hipóxia.
ABSTRACT
Hypoxia has emerged as a key determinant of osteogenesis. HIF-1
is the
transcription factor mediating hypoxia responses that include induction of VEGF and
related bone induction. Inflammatory signals antagonize bone repair via the NF-
B
pathway. The present investigation explored the functional relationship of hypoxia
(HIF-1
function) and inflammatory signaling (NF-
B) in stem like and
osteoprogenitor cell lines. The potential interaction between HIF-1
and NF-
B
signaling was explored by co-transfection studies in hFOB with p65, HIF-1
and
9x-HRE-luc or HIF-1
target genes reporter plasmids. Nuclear cross-talk was directly
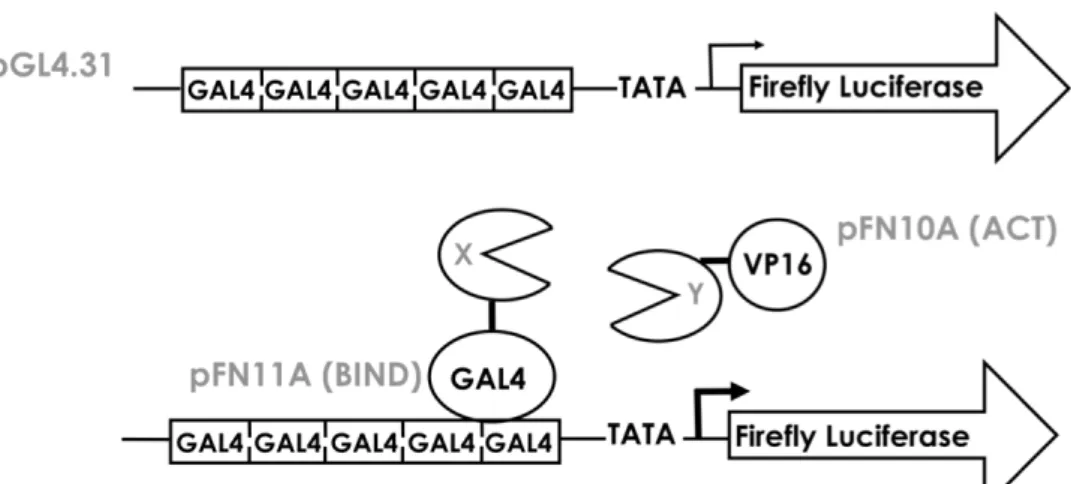
tested using the mammalian Gal4/VP16 two-hybrid, and confirmed by
co-immunoprecipitation/western blotting assays. The results show that inflammatory
stimulation (TNF-
treatment) causes a marked inhibition of HIF-1
function at the
HRE in all cell lines studied. Also, co-transfection with p65 expression vector leads to
reduced hVEGFp transcription after DFO-induced hypoxia. However, TNF-
treatment or NF-
B expression from encoding virus or plasmid had little effect on
HIF-1
mRNA or protein levels nor did HIF- 1
nuclear translocation change with
treatment. The functional interaction of Gal4-HIF-1
and VP16-p300 fusion proteins
is effectively blocked by expression of p65 in a dose dependent manner. It was
concluded that NF-
B-mediated inflammatory signaling is able to block HIF-1
transactivation at HRE-encoding genes by direct competition for p300 binding at the
promoter. Inflammation may influence the stem cell niche and tissue regeneration by
influencing cellular responses to hypoxia.
LISTA DE ABREVIATURAS
ACTB – Beta actin
ANG-1 – Angiopoietin 1
ANG-2 – Angiopoietin 2
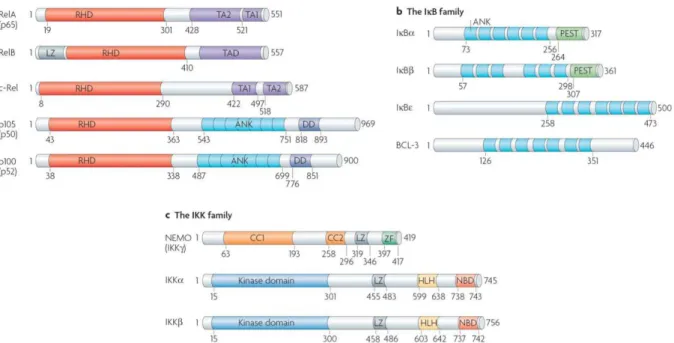
ANK – Ankyrin-repeat motifs
ARNT – Aryl hydrocarbon receptor nuclear translocator
ATM – Ataxia telangiectasia mutated checkpoint kinase
BAFF – B-cell activating factor
bHLH – Basic helix-loop-helix domain
BMP – Bone morphogenetic protein
BSA – Bovine serum albumin
C-TAD – Carboxi-terminal Activation Domain
CAD – Carboxi-terminal Activation Domain
CD14 – Cluster of differentiation 14
CH1 – Cysteine-histidine-rich zinc finger motif
CK2 – Casein kinase-II
COX2 – Ciclooxigenase 2
CSC – Cancer stem cells
CX3CR1 – CX3C chemokine receptor 1
CXCR4 – CXC chemokine receptor 4
DFO – Desferrioxamine
EGF – Epidermal growth factor
eNOS – Endothelial nitric oxide synthase
ERK – Extracellular signal-regulated kinase
FAK – Focal adhesion kinase
FBS – Fetal bovine serum
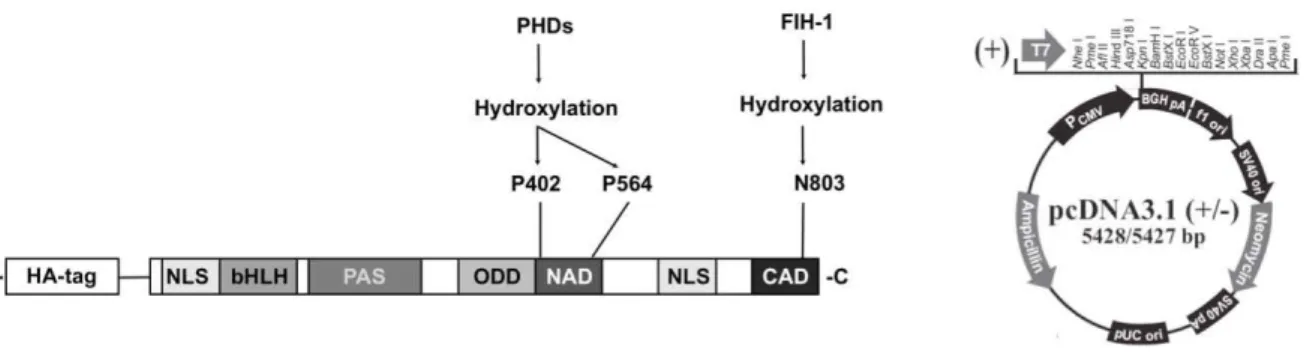
FIH-1 – Factor inhibiting HIF-1
FITC – Fluorescein isothiocyanate
HA – Hemagglutinin
HAT – Histone acetyltransferases
HDAC – Histone deacetylases
HET – Heterozygous
hFOBs – Human fetal osteoblasts
HIF-1
– Hypoxia inducible factor 1
hMSCs – Human mesenchymal stem cells
HRE – Hypoxia responsive element
HRP – Horseradish peroxidase
ID – Inhibitory domain
IGF-1 – Insulin-like growth factor 1
IGF-2 – Insulin-like growth factor 2
IKK – Inhibitor of
B kinase
IL-1 – Interleukin-1
IL-4 – Interleukin 4
IL-8 – Interleukin-8
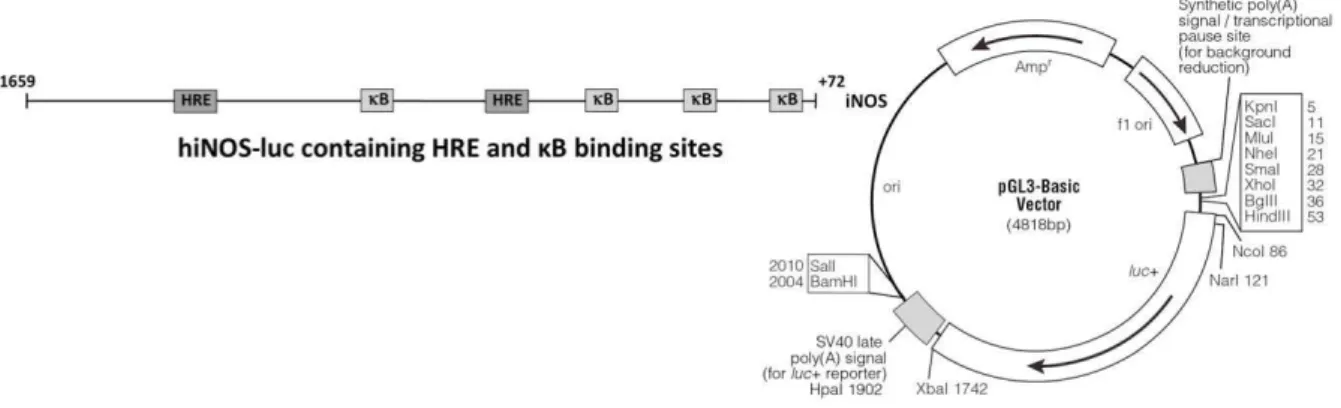
iNOS – Inducible nitric oxide synthase
I
B
-SR – I
B
super repressor
LAR II – Luciferase Assay Reagent II
LMP1 – Latent membrane protein 1
LMP1 – Latent membrane protein-1
LOX – Lysyl oxidase
LPS – Lipopolysaccharide
LT
R – Lymphotoxin-
receptor
MAPK – Mitogen-activated protein kinase
ml – milliliter
MMP-2 – Matrix metalloproteinase 2
MMP-2 – Matrix metalloproteinase 9
MOI – Multiplicity of infection
mTOR – Mammalian target of rapamycin
N-TAD – Amino-terminal Activation Domain
N803 – Asparaginyl residue 803
NEMO – NF-
B essential modifier
NF-
B: Nuclear factor kappa B
ng – nanogram
NGF – Nerve growth factor
NIK – NF-
B inducing kinase
NO – Nitric oxide
ODD – Oxygen-dependent domain
PAI-1 – Plasminogen activator inhibitor 1
PAS – Per-ARNT-Sim domain
PBS – Phosphate buffered saline
PCR – Polymerase Chain Reaction
PDGF-
– Platelet-derived growth factor
PHD – Prolyl hydroxylase
PI-3k/AKT – Phosphatidylinositol-3 kinase / Serine-threonine protein kinase pathway
PKA – Protein kinase A
PKC – Protein kinase C
pRL-TK –
Renilla
luciferase reporter that contains the Herpes simplex virus
Thymidine Kinase
Pro 564 – Proline residue 564
Pro402 – Proline residue 402
pVhl – Von Hippel-Lindau protein
Rel homology domain – RHD
RIPA – Radio-immunoprecipitation assay
ROS – Reactive oxygen species
RT-PCR – Reverse transcriptase – Polymerase chain reaction
SDF-1
– Stromal-derived factor 1
SDS-PAGE – Sodium dodecyl sulfate – Polyacrylamide gel electrophoresis
SNIP1 – Smad nuclear-interacting protein 1
SV40 – Simian vacuolating virus
TBS – Tris-buffered saline
TNF-
– Tumor necrosis factor
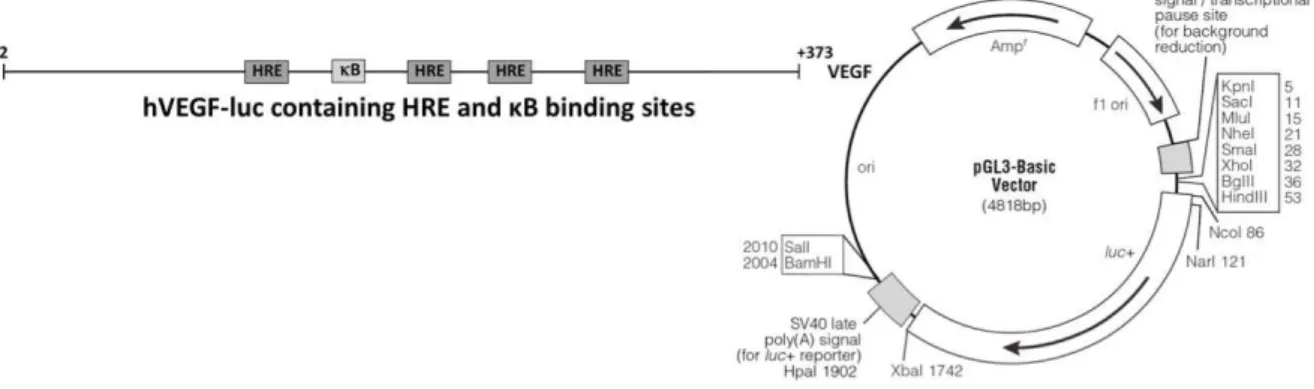
VEGF – Vascular endothelial growth factor
µg – microgram
SUMÁRIO
Resumo
Abstract
Lista de Abreviaturas
Introdução
14
Revisão da literatura
25
Objetivos
47
Materiais e métodos
49
Resultados
59
Discussão
90
Conclusões
97
Perpectivas futuras
100
Referências Bibliográficas
101
O fator induzido por hipóxia 1 alfa (Hypoxia Inducible Factor, HIF-1
) foi
inicialmente identificado como um fator de transcrição responsável pela regulação do
gene da eritropoetina em resposta à uma diminuição de oxigênio nos tecidos renais
(WENGER et al., 2005). O complexo HIF consiste de uma de três sub-unidades alfa
(HIF-1
, HIF-2
ou HIF-3
) que se ligam ao Translocador Nuclear Receptor Aril
Hidrocarboneto (Aryl Hydrocarbon Receptor Nuclear Translocator, ARNT), também
conhecido como subunidade beta do fator 1 induzível por hipóxia HIF-1
(WANG et
al., 2007a). Geralmente, o HIF-1
é constitutivamente expresso e seus níveis não
variam de acordo com a disponibilidade de oxigênio, ao contrário das subunidades
alfa que são estritamente reguladas em resposta à hipóxia (HIROTA; SEMENZA,
2005). As variações na concentração de oxigênio não são diretamente reguladas
pelo HIF-1
e sim por uma classe de dioxigenases dependentes de ferro e de
2-oxoglutarato. Dois tipos de sensores do nível de oxigênio estão envolvidos na
regulação do HIF-1
: as prolil hidroxilases (PHDs) e a asparaginil hidroxilase
(SCHIPANI et al., 2009). Na presença de níveis normais de oxigênio (normóxia), as
PHDs 1, 2 e 3 hidroxilam dois resíduos de prolina (Pro402 e Pro564) na porção
amino-terminal (N-terminal transactivation
domain, N-TAD) do domínio de
degradação dependente de oxigênio (Oxygen-dependent Domain, ODD). Essa
hidroxilação promove a interação do HIF-1
com a proteína do gene supressor de
tumor von Hippel-Lindau (von Hippel-Lindau protein, pVHL), que é o componente de
reconhecimento da ligase de ubiquitina E3. O HIF-1
é então marcado com cadeias
de poliubiquitina e será degradado pelo proteassomo. O segundo sensor de oxigênio
é uma asparaginil hidroxilase conhecida como Fator Inibidor do HIF-1 (Factor
Inhibiting HIF-1, FIH-1). Essa enzima hidroxila um resíduo de asparagina (N803) na
porção carboxi-terminal (C-terminal transactivation domain, C-TAD) do domínio ODD
do HIF-1
. Essa modificação covalente bloqueia a interação do C-TAD com os
co-ativadores transcricionais, como CBP e p300 (SCHIPANI et al., 2009).
(RCGTG) na região promotora dos genes alvo do HIF-1
. Em células bem
oxigenadas, os dois sensores de oxigênio, PHD e FIH, que regulam a destruição e
atividade do HIF-1
, respectivamente, asseguram a repressão da via do HIF-1
em
células bem oxigenadas (SCHIPANI
et al.,
2009). A
figura 1 mostra o HIF-1
em
condições de normóxia e hipóxia (SEMENZA, 2004).
Acredita-se que a hipóxia seja o principal estímulo responsável pelo início da
cascata angiogênica durante o desenvolvimento e após o traumatismo ósseo. Os
osteoblastos estão idealmente localizados no osso de maneira que possam sentir a
tensão de oxigênio e responder à hipóxia ativando a via do HIF-1
(WANG et al.,
2007b). Utilizando uma abordagem genética para determinar os efeitos celulares e
moleculares de ganho ou perda da função do HIF-1
por mutagênese condicional
em osteoblastos de camundongo, esses autores demonstraram que a
superexpressão do HIF-1
em osteoblastos de camundongo por meio da truncagem
do Vhl resultou em aumento profundo na angiogênese e osteogênese. Ao contrário,
a truncagem do HIF-1
nos osteoblastos produziu o fenótipo reverso, ou seja, ossos
mais finos e menos vascularizados, o que demonstra a importância da via do HIF-1
na formação óssea guiada por osteoblastos (WANG et al., 2007a). O mesmo grupo
também demonstrou que a ativação farmacológica da via do HIF-1 por meio do
DFO induziu uma robusta resposta angiogênica acompanhada de uma subsequente
resposta osteogênica in vivo (WAN et al., 2008).
Atualmente, mais de 100 genes alvo do HIF-1
foram identificados, incluindo
genes relacionados à inflamação como a cicloxigenase-2 (COX-2), citocinas como o
fator de necrose tumoral (Tumor Necrosis Factor alpha, TNF-
) e a interleucina 8
(Interleukin-8, IL-8). Então, além de facilitar a adaptação, a hipóxia pode também
afetar os processos inflamatórios (OLIVER et al., 2009).
Gene product
1B-Adrenergic receptor
Adrenomedullin Aldolase A (ALDA) Atrial natriuretic peptide Carbonic anhydrase 9 CD18
Ceruloplasmin C-MET
Connective tissue growth factor CYP3A6
CXCR4 DEC1 DEC2
Ecto-5_-nucleotidase (CD73) Endocrine gland-derived VEGF Endoglin Endothelin-1 Enolase 1 ENOS Erythropoietin ETS-1
Glucose transporter 1 (GLUT1) Glyceraldehyde-3-phosphate dehydrogenase
Glucose regulated protein, 94-kDa (GRP94)
Heme oxygenase-1
HIF-1_ prolyl hydroxylase PHD3 (EGLN3)
HIF-1_ prolyl hydroxylase PHD2 (EGLN1)
HGTD-P Integrin 2
Intestinal trefoil factor
Lactate dehydrogenase A (LDHA) Lactase
Leptin
Membrane type-1 matrix metalloproteinase
Multi-drug resistance 1 (ABCB1) Myeloid cell factor 1 (MNL1) Nitric oxide synthase 2 NIP3
NUR77
p35srj (CITED2)
Phosphoglycerate kinase 1
6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3)
6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase-4 (PFKFB4)
Plasminogen activator inhibitor 1 Procollagen prolyl-4-hydroxylase (I) ROR
Stromal-derived factor 1 (SDF-1) Telomerase (TERT)
Transferrin
Transferrin receptor
Transforming growth factor 3 Vascular endothelial growth factor (VEGF)