Article
J. Braz. Chem. Soc., Vol. 24, No. 6, 1018-1026, 2013. Printed in Brazil - ©2013 Sociedade Brasileira de Química 0103 - 5053 $6.00+0.00
A
*e-mail: [email protected] , [email protected]
Theoretical Studies on High Energetic Density Polynitroimidazopyridines
Ming Lu* and Guozheng Zhao*
School of Chemical Engineering, Nanjing University of Science & Technology, Nanjing, Jiangsu, 210094, P. R. China
Teoria do funcional da densidade (DFT) foi empregada no estudo de poli-nitroimidazopiridinas em nível B3LYP/6-31+G(d). A velocidade (D) e a pressão de detonação (P) foram avaliadas utilizando as equações Kamlet-Jacobs (K-J) baseadas na densidade molecular teórica (ρ) e calor de formação (HOF). A estabilidade térmica dos compostos foi investigada através dos cálculos das energias de dissociação das ligações (BDE) em nível B3LYP/6-31+G(d) irrestrito. Alguns compostos apresentam altas densidades (ca. 1,95 g cm-3) e bons desempenhos. Os resultados das
simulações revelam que duas das moléculas atuam similarmente ao 1,3,5,7-tetranitro-1,3,5,7-tetrazocano (HMX), e outras duas moléculas podem ser potenciais candidatas a compostos de alta densidade de energia (HEDCs). Estes resultados fornecem informações básicas para o planejamento molecular de novos compostos de alta densidade energética.
Density functional theory (DFT) was employed to study polynitroimidazopyridines at the B3LYP/6-31+G(d) level. Detonation velocity (D) and detonation pressure (P) were evaluated using Kamlet-Jacobs (K-J) equations based on the theoretical molecular density (ρ) and heat of formation (HOF). Thermal stability of the title compounds was investigated by calculating the bond dissociation energies (BDE) at the unrestricted B3LYP/6-31+G(d) level. Some compounds have high densities (ca. 1.95 g cm-3) and good performance. Simulation results reveal that two molecules
perform similarly to 1,3,5,7-tetranitro-1,3,5,7-tetrazocane (HMX), and other two molecules may be potential candidates of high energy density compounds (HEDCs). These results provide basic information for molecular design of novel high energetic density compounds.
Keywords: density functional theory, polynitroimidazopyridines, heat of formation, detonation
property, thermal stability
Introduction
Energetic materials are more and more widely applied due to their high energy density.1-4 In order to meet the
requirements of future military and space applications, continuous efforts have been made to develop new materials having good thermal stability, impact and shock insensitivity, better performance, economic and environmentally friendly syntheses.5-7 These properties
are essential to improve personnel safety and to reduce warhead vulnerability problems.But these requirements are somewhat reciprocally exclusive, with improved insensitivity bringing inferior performance and vice versa. Therefore, the need for more energetic compounds with better stability and lower sensitivity is one of the main goals of energetic material research.8,9
Nitramine compounds, an important class of organic explosives, have received much attention as an energetic material due to an advantageous combination of density, heat of formation and oxygen balance. Nitramine compounds are well known by their high positive heat of formation as well as good thermal stability.10,11
These properties reveal a high performance of these energetic materials. The most prominent members of this class are 1,3,5-trinitro-1,3,5-triazinane (RDX)12-15 and
1,3,5,7-tetranitro-1,3,5,7-tetrazocane (HMX).16-19 CL-20
[2,4,6,8,10,12-hexanitrohexaazaisowurtzitane (HNIW)] is another new nitramine explosive, which has six N–NO2
groups in its polycyclic structure, resulting in an increase in both density and detonation properties.20-25 CL-20 is
reported as an attractive high thermally stable explosive with a decomposition temperature at 228 oC. It can offer
high velocity of detonation (9.38 km s-1) and heat of
performance.
This study was motivated by, and based on, the concept of new nitramine explosives containing pyridine. Reacting with formaldehyde, pyridine-2,3,5,6-tetraamine yields a precursor with two imidazole rings, providing more N–H and C–H sites for introducing nitro substituents, and thus generating
molecular structure, with or without using experimental measurements, has been a prime requirement in the field of explosive research and development.27-30 In this work, the full
geometrical optimizations of nitramine compounds containing a benzene ring at the DFT-B3LYP/6-31+G(d) level were performed. The structure-property studies were performed
to find out potential candidates for high energy density compounds (HEDCs).4 The B3LYP/6-31G(d) method of
density functional theory was used to study the thermodynamic properties and detonation properties of cage-HMX.31
To date, information on the relationships between structure and property of polynitroimidazopyridines was very sparse, there was no systematic survey covering these compounds. In the present study, the molecular geometries and electronic structure were obtained with density functional theory (DFT) method. Based on optimized geometries, the molecular volume (V) and theoretical density (ρ) were calculated using Monte-Carlo
method. The most important detonation properties, such as detonation velocity (D) and detonation pressure (P), were estimated by using the Kamlet-Jacobs (K-J) equations. Through calculations of bond dissociation energies (BDE), the thermal stability was studied. These results provide theoretical support for the molecular design of novel high energetic density compounds.
Methodology
Computations were performed with Gaussian 03 package at B3LYP32-34 method with 6-31+G(d) basis
set.35 The geometric parameters were allowed to be
optimized, and no constraints were imposed on molecular structure during optimization process. Vibrational frequencies were calculated for the optimized structures to enable the natural characterization of stationary points, zero-point energy (ZPE) and thermal correction (HT). All of optimized structures were characterized to be true local energy minima on potential energy surfaces without imaginary frequencies.
Detonation velocity and detonation pressure are the most important parameters to evaluate detonation characteristics of energetic materials. For the explosives
with CHNO elements, the Kamlet-Jacobs empirical equations were used to determine the parameters.36
P = 1.558 NM1/2Q1/2ρ2 (1)
D = 1.01 (NM1/2Q1/2)1/2 (1 + 1.30 ρ) (2)
where P is the detonation pressure in GPa, D is the detonation velocity in km s-1, N is the number of moles of
gaseous detonation products per gram of explosive, M is the average molecular weight of the gaseous products, Q is the energy of explosion in J per gram of explosive and ρ is the crystal density in g cm-3. N, M and Q are decided according to the largest exothermic principle,37 i.e., for
the explosives with CHNO elements, all N atoms convert into N2, the O atoms form H2O with H atoms first and the
remainder forms CO2 with C atoms. The remaining C atoms
will exist in the solid state if the O atoms do not satisfy full oxidation of C atoms. The remaining O atoms will exist in O2 if O atoms are in excess.
The chemical energy of the detonation reaction Q was calculated as the difference between the heats of formation (HOFs) of products and reactants. In this work, HOFs were calculated using density functional method with the help of the following equations:
aC(s) + 0.5bH2(g) + 0.5cO2(g) + 0.5dN2(g) =
CaHbOcNd(g) (3)
The HOF of CaHbOcNd in gas phase (∆Hf(g)) is readily
obtained from equation 3 with the experimental HOFs of C(s), H2(g), O2(g) and N2(g), and the calculated enthalpies
of all species.
energies of the product and reactant after zero-point energy correction. The expressions for the homolysis of the A-B bond (equation 4) and for calculating its BDE equation 5 are shown as follows: 42
A-B(g) → A•(g) + B•(g) (4)
BDE(A-B)ZPE = E(A•)ZPE + E(B•)ZPE − E(A-B)ZPE (5)
where A-B stands for neutral molecules and A• and B• for
the corresponding product radicals after bond dissociation; BDE(A-B) is the BDE of bond A-B; E(A-B)ZPE, E(A•)ZPE and E(B•)ZPE are zero-point energy corrected total energies of the parent compound and corresponding radicals, respectively.
Results and Discussion
Optimized structures
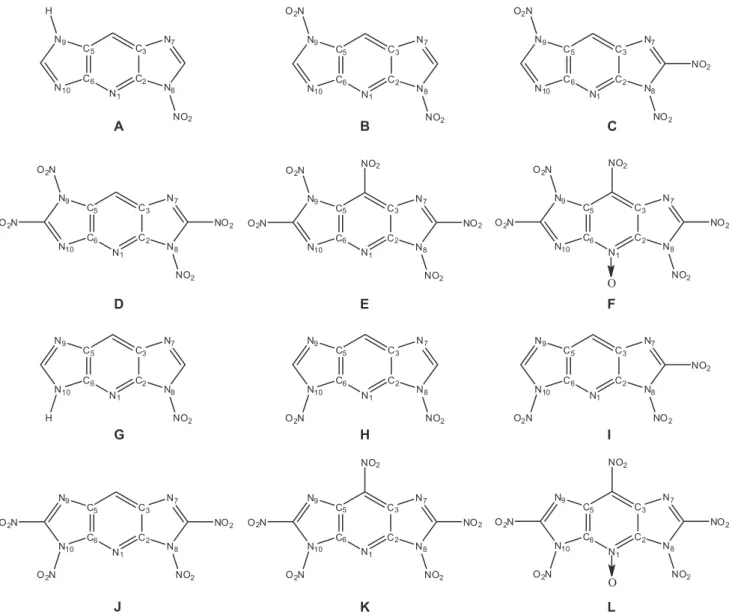
Optimized bond lengths of polynitroimidazopyridines are tabulated in Table 1. The pyridine rings in
polynitro-to the large steric hindrance effect of –NO2. The same
results are obtained for molecules G < H < I < J < K < L. The corresponding bond lengths of molecules E and F are close to each other. Some discrepancies are obviously raised by the formation of N→O coordination bond. The
lengths of all N−NO2 bonds in polynitroimidazopyridines
are longer than the usual N−N bond lengths (1.35-1.40 Å)
in nitramines.43 The N−NO
2 bonds of molecules E and K
are shorter than those of molecules F and L, respectively. Obviously, this change is caused by the introduction of the O coordination atom.
Density
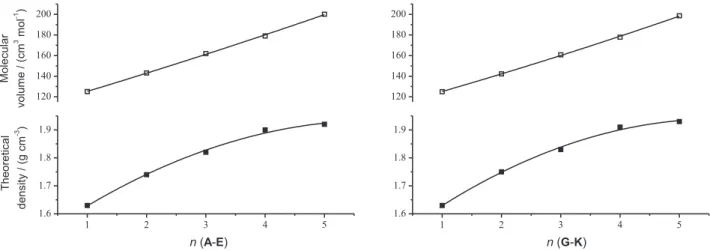
In the present study, single-point molecular volume calculations at B3LYP/6-31+G(d) were performed based on optimized geometries. The densities provide some clue about the explosive character of these molecules. The relationship between V, ρand the number of nitro groups (n) can be expressed as shown in Figure 2. The largest value and the smallest one are 1.95 and 1.63 g cm-3, respectively. From
A to E, it is clear that an increase in density is observed
Table 1. Selected bond lengths (Å) of polynitroimidazopyridines computed at B3LYP/6-31+G(d) level
Bond length / Å
A B C D E F
N1–C2 1.317 N1–C2 1.320 N1–C2 1.319 N1–C2 1.319 N1–C2 1.318 N1–C2 1.361
C3–N7 1.395 C3–N7 1.393 C3–N7 1.385 C3–N7 1.385 C3–N7 1.380 C3–N7 1.377
C2–N8 1.414 C2–N8 1.409 C2–N8 1.404 C2–N8 1.402 C2–N8 1.399 C2–N8 1.372
C5–N9 1.382 C5–N9 1.400 C5–N9 1.399 C5–N9 1.406 C5–N9 1.400 C5–N9 1.388
C6-N10 1.382 C6-N10 1.394 C6-N10 1.391 C6-N10 1.391 C6-N10 1.390 C6-N10 1.358 Bond length / Å
G H I J K L
N1–C2 1.324 N1–C2 1.325 N1–C2 1.324 N1–C2 1.324 N1–C2 1.323 N1–C2 1.359
C3–N7 1.398 C3–N7 1.395 C3–N7 1.387 C3–N7 1.388 C3–N7 1.383 C3–N7 1.383
C2–N8 1.409 C2–N8 1.406 C2–N8 1.402 C2–N8 1.398 C2–N8 1.396 C2–N8 1.370
C5–N9 1.388 C5–N9 1.395 C5–N9 1.395 C5–N9 1.388 C5–N9 1.383 C5–N9 1.383
with an increase in the number of nitro groups. The same results were obtained for molecules G, H, I, J and K. The introduction of a nitro group increases the density of molecules and therefore has a significant contribution to the detonation velocity and detonation pressure. Molecules D, E, F, J, K and L have a density of above 1.90 g cm-3 when it is successfully synthesized. ρ is the essential
factor in determining the detonation properties of energetic compounds.26 According to Kamlet-Jacobs semi-empirical
equations, D increases with increasing ρ for most energetic
compounds. Also, P varies with the square of ρ, when ρ
is greater than one.
Heat of formation
Heat of formation reflects the energy content of a compound. High positive HOF is usually required for an effective energetic material. The zero-point energies (ZPE), thermal correction to enthalpy and electronic energies calculated at the B3LYP/6-31+G(d) level for polynitroimidazopyridines are listed in Table 2. It is evident from the data listed in Table 2 that all HOFs of the title compounds are quite large positive values. HOFs of RDX and HMX computed at the B3LYP/6-31+G(d) level are 216.76 and 251.67 kJ mol-1, respectively. For
molecules A and B, G and H, each nitro group addition will increase HOF by 318.68 and 340.28 kJ mol-1,
respectively. But for molecules B and C, H and I, each nitro group addition will increase HOF by 296.52 and 296.18 kJ mol-1, respectively. It indicates that the value
of HOF relates to the nature of C−NO2 and N–NO2. The
space orientations of nitro groups also affect HOFs of the title compounds. For the isomers with the same number of –NO2 groups, the values of HOF are slightly different,
indicating that HOF is a little influenced by the position of –NO2 group. As a whole, according to the number and
the relative position of the nitro groups, the relative HOF order of molecules can be distinguished, which is useful for evaluating the relative thermal stability of nitramine compounds.
Detonation properties
The detonation velocity and detonation pressure of molecules are computed by Kamlet-Jacobs empirical equations on the basis of the theoretical densities and calculated gas phase heats of formation, which are the important parameters to evaluate explosion performance of energetic materials. Table 3 shows the predicted detonation properties of polynitroimidazopyridines. Because detonation pressure and detonation velocity are calculated by HOF of the gas state, not of the crystal, Figure 2. Correlations between molecular volume (V), theoretical density (ρ) and the number of nitro groups (n) for polynitroimidazopyridines
Table 2. Calculated electronic energies (E0), zero-point energies (ZPE),
thermal correction to enthalpy (HT) and gas phase heats of formation
(HOF)
Molecule E0 / a.u. ZPE / a.u. HT / a.u.
HOF / (kJ mol-1)
A −747.887724 0.125526 0.011301 176.57
B −952.358263 0.127119 0.013869 495.25
C −1156.837087 0.128710 0.016665 791.77
D −1361.312357 0.130218 0.019550 1097.63
E −1565.788593 0.131711 0.021627 1400.95
F −1640.932287 0.134883 0.023748 1499.34
G −747.893184 0.125758 0.011235 162.64
H −952.355340 0.126913 0.013973 502.92
I −1156.834296 0.128478 0.016773 799.10
J −1361.311599 0.129955 0.019621 1099.62
K −1565.792730 0.131570 0.022712 1390.08
the calculated detonation properties of the nitramine compounds have some deviation,44 but the results are still
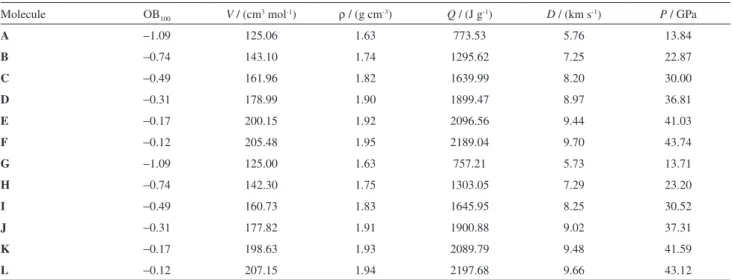
reliable and meaningful. It can be found from Table 3 that all polynitroimidazopyridines have good detonation properties (Q = 757.21-2197.68 J g-1, D = 5.73-9.70 km s-1, P = 13.71-43.74 GPa). Meanwhile, with the number of nitro groups increasing from one to five, Q, D and P of the corresponding compounds increase. Molecule F is calculated to have the highest D and P values among polynitroimidazopyridines. In terms of the predicted detonation parameters, the most powerful explosives among polynitroimidazopyridines are molecules F and L.
As for the isomers with the same oxygen balance (OB100),
no conspicuous discrepancy of their respective Q, D and P is found. As a whole, Q, D and P increase with the increasing number of –NO2 groups. Figure 3 and Table 4 present the
relationships between D, P and n. This may show good group additivity on detonation properties and support the claim that
introducing more nitro substituents into a molecule usually helps to increase its detonation performance.37
Compared with the famous nitramine explosive RDX (1,3,5-trinitro-1,3,5-triazinane) (ρ = 1.82 g cm-3, D = 8.75 km s-1, P = 34.70 GPa),45,46 they have better detonation performance when the number of nitro groups is not less than 4, indicating that they are potential energetic compounds. Calculation results of detonation velocity and detonation pressure for nitramine compounds indicate that molecules D, J, E, K, F and L outperform RDX, molecules D and J perform similarly to HMX (1,3,5,7-tetranitro-1,3,5,7-tetrazocane) (ρ = 1.92 g cm-3, D = 8.96 km s-1,
P = 35.96 GPa).45,46 According to the energy criterion for HEDC, i.e., ρ ≥ 1.90 g cm-3, D ≥ 9.0 km s-1, and P≥ 40.0 GPa, it is found in Table 3 that molecules E, K, F and L satisfy the requirements as novel high energy density compounds. Therefore, in the molecular design, detonation properties by changing the substituted group
I −0.49 160.73 1.83 1645.95 8.25 30.52
J −0.31 177.82 1.91 1900.88 9.02 37.31
K −0.17 198.63 1.93 2089.79 9.48 41.59
L −0.12 207.15 1.94 2197.68 9.66 43.12
V: molecular volume; ρ: theoretical molecular density; Q: energy of explosion; D: detonation velocity; P: detonation pressure.
could be adjusted. Therefore, the above prediction indicates that polynitroimidazopyridines appear to be promising candidates comparable to nitramine explosives RDX and HMX.
Thermal stability
The relationship between the impact sensitivity and electronic structures of polynitroimidazopyridines can be established by the charge analysis of thenitro group.47
Nitro compounds are very strong electron acceptors and have a strong ability to attract electrons. Such ability can be represented by the net charges of the nitro group. The lower the negative charge on the nitro group, the lower the electron attraction ability, and therefore, the more stable the nitro compound. In the present study, the charge on the nitro group (−QNO2) is considered for its correlation to impact sensitivity:
QNO2= QN + QO1 + QO2 (6)
The charge on the nitro group (−QNO2) is calculated by the sum of atomic charges on the nitrogen (QN) and oxygen (QO1 and QO2) atoms of the nitro group. −QNO2 can be regarded as the criterion for estimating the impact sensitivities. Based on the highest −QNO2 value, the probable decreasing order is as follows: molecule L (0.245) > molecule F (0.234) > molecule E (0.136)> molecule K (0.118). This shows that molecule L is more sensitive than the other molecules.
Studies of BDE provide useful information for understanding the stability of polynitroimidazopyridines.
The stability of compounds is affected by bond dissociation energies, so the weakest bonds (N–N bonds that are out of ring) were selected as the breaking bond based on the bond overlap populations to calculate BDE at B3LYP/6-31+G(d) level. The values of bond dissociation energies are listed in Table 5. The calculated BDEZPE values indicate the relative stability of energetic
materials. Variations of BDEZPE for N–NO2 are in the range
of 4.31-132.73 kJ mol-1. The initial step should be via
N−NO2 cleavage in thermal decomposition. The BDEZPE
value of molecule B (132.73 kJ mol-1) is the largest while molecule L is the smallest (4.31 kJ mol-1) indicating
that the former is more stable than the latter. Compared with BDEZPE values of RDX (141.22 kJ mol-1) and HMX
(146.61 kJ mol-1) computed at the B3LYP/ 6-31+G(d)
level, polynitroimidazopyridines are less stable than RDX and HMX.
By analyzing the structures of these compounds, it is easy to find that nitramine compounds have symmetric structures. Therefore, molecules B, D, H and J have higher BDEZPE than molecules A, C, G and I, respectively. This
indicates that a symmetric structure is very useful for improving thermal stability. The symmetry can delocalize the π electron cloud density of system, but the five-membered
rings of these compounds have larger tension, leading to the BDEZPE decrease of polynitroimidazopyridines. Repulsion
is an important role in the stability of the title compounds. Take molecule L as an example, the repulsion between the neighboring nitro group rotates the oxygen atoms from the molecular plane and makes the value of BDEZPE decrease.
This shows that structures of these compounds have a great influence on the thermal stability.
Table 4. Correlation equations between V, ρ, D, P and the number of nitro groups (n) for the title compounds
A-E G-K
V = 108.23 + 16.72n + 0.32n2 R2 = 0.9988 V = 108.90 + 15.84n + 0.41n2 R2 = 0.9991
ρ = 1.49 + 0.15n – 0.01n2 R2 = 0.9928 ρ = 1.48 + 0.16n – 0.01n2 R2 = 0.9940
D = 4.09 + 1.86n – 0.16n2 R2 = 0.9979 D = 3.99 + 1.95n – 0.17n2 R2 = 0.9972
P = 3.44 + 11.09n – 0.71n2 R2 = 0.9989 P = 2.83 + 11.68n – 0.78n2 R2 = 0.9992
V: molecular volume (cm3 mol-1); ρ: theoretical molecular density (g cm-3); D: detonation velocity (km s-1); P: detonation pressure (GPa); R2: correlation
coefficient.
Table 5. Bond dissociation energies (BDE) of the weakest bonds for polynitroimidazopyridines computed at the B3LYP/6-31+G(d) level
Compound A B C D E F
Bond N8–NO2 N8–NO2 N9–NO2 N8–NO2 N9–NO2 N9–NO2
BDEZPE / (kJ mol-1) 125.22 132.73 86.27 90.19 76.05 16.22
Compound G H I J K L
Bond N8–NO2 N8–NO2 N10–NO2 N10–NO2 N10–NO2 N10–NO2
The full geometrical optimizations of polynitroimidazo-pyridines were performed using density functional theory at the B3LYP/6-31+G(d) level, without any symmetry restriction. The detailed structure-property studies were performed on these molecules to achieve energetic performance for the first time. Stability correlations are established for the title compounds by analyzing bond dissociation energies. For the polynitroimidazopyridines, with the increase in the number of nitro groups, the volume, density, detonation velocity and detonation pressure increase. Calculation results of detonation velocity and detonation pressure for the compounds indicate that molecules D, J, E, K, F and L outperform RDX, and molecules D and J perform similarly to HMX. According to the calculated BDE, the N–NO2 bond is the trigger
bond during the thermolysis initiation process. Molecules E and K essentially satisfy the quantitative criteria of energetics and stability as HEDCs. These results provide theoretical support for the molecular design of novel high energetic density compounds and experimental synthesis.
Acknowledgments
This work was supported by the NSAF Foundation of the National Natural Science Foundation of China and China Academy of Engineering Physics (Grant No 11076017).
References
1. Gutsev, G. L.; Weatherford, C. A.; Johnson, L. E.; Jena, P.;
J. Comput. Chem.2012, 33, 416.
2. Bushuyev, O. S.; Brown, P.; Maiti, A.; Gee, R. H.; Peterson, G. R.; Weeks, B. L.; Hope-Weeks, L. J.; J. Am. Chem. Soc.
2012, 134, 1422.
3. Windler, G. K.; Zhang, M. X.; Zitterbart, R.; Pagoria, P. F.; Vollhardt, K. P. C.; Chem. Eur. J.2012, 18, 6588.
4. Zhao, G. Z.; Lu, M.; J. Mol. Model.2012, 18, 2443.
5. Elbeih, A.; Jungova, M.; Zeman, S.; Vavra, P.; Akstein, Z.;
Propelllants Explos. Pyrotech.2012, 37, 329.
13. Qiu, H.; Stepanov, V.; Di, S. A. R.; Chou, T.; Lee, W. Y.;
J. Hazard. Mater.2011, 185, 489.
14. Joseph, M. D.; Jangid, S. K.; Satpute, R. S.; Polke, B. G.; Nath, T.; Asthana, S. N.; Rao, A. S.; Propelllants Explos. Pyrotech.2009, 34, 326.
15. Podeszwa, R.; Rice, B. M.; Szalewicz, K.; Phys. Chem. Chem. Phys.2009, 11, 5512.
16. Zhou, T. T.; Huang, F. L.; J. Phys. Chem. B2011, 115, 278.
17. Konek, C. T.; Mason, B. P.; Hooper, J. P.; Stoltz, C. A.; Wilkinson, J.; Chem. Phys. Lett.2010, 489, 48.
18. Qiu, L.; Zhu, W. H.; Xiao, J. J.; Xiao, H. M.; J. Phys. Chem. B
2008, 112, 3882.
19. Glascoe, E. A.; Zaug, J. M.; Burnham, A. K.; J. Phys. Chem. A
2009, 113, 13548.
20. Maksimowski, P.; Fabijanska, A.; Adamiak, J.; Propelllants Explos. Pyrotech.2010, 35, 353.
21. Bayat, Y.; Hajimirsadeghi, S. S.; Pourmortazavi, S. M.; Org. Process Res. Dev.2011, 15, 810.
22. Lin, C. P.; Chang, C. P.; Chou, Y. C.; Chu, Y. C.; Shu, C. M.;
J. Hazard. Mater.2010, 176, 549.
23. Sorescu, D. C.; Rice, B. M.; J. Phys. Chem. C2010, 114, 6734. 24. Saint, M. S.; Marre, S.; Guionneau, P.; Cansell, F.; Renouard, J.; Marchetto, V.; Aymonier, C.; Chem. Eur. J. 2010, 16, 13473. 25. Haycraft, J. J.; J. Chem. Phys.2009, 131, 214501.
26. Sikder, A. K.; Sikder, N.; J. Hazard. Mater.2004, 112, 1. 27. Tuerker, L.; Varis, S.; J. Energetic Mater.2013, 31, 203.
28. Witko, E. M.; Korter, T. M.; J. Phys. Chem. A2012, 116, 6879. 29. Turker, L.; Atalar, T.; Gumus, S.; Camur, Y.; J. Hazard. Mater.
2009, 167, 440.
30. Molt, R. W.; Bartlett, R. J.; Watson, T.; Bazante, A. P.; J. Phys. Chem. A2012, 116, 12129.
31. Zhao, G. Z.; Lu, M.; Struct. Chem.2013, 24, 139.
32. Becke, A. D.; J. Chem. Phys.1992, 97, 9173.
33. Lee, C.; Yang, W.; Parr, R. G.; Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785.
34. Becke, A. D.; J. Chem. Phys.1993, 98, 5648.
J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q. K.; Morokuma, D. K.; Malick, A. D.; Rabuck, K.; Raghavachari, J. B.; Foresman, J. C.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A.; Gaussian 03, Gaussian, Inc.; Pittsburgh, PA, USA, 2003.
36. Kamlet, M. J.; Jacobs, S. T.; J. Chem. Phys.1968, 48, 23. 37. Wang, G. X.; Gong, X. D.; Liu, Y.; Xiao, H. M.; Int. J. Quantum
Chem. 2010, 110, 1691.
38. Xu, X. J.; Xiao, H. M.; Ju, X. H.; Gong, X. D.; Zhu, W. H.;
J. Phys. Chem. A2006, 110, 5929.
39. Qiu, L.; Xiao, H. M.; Gong, X. D.; Ju, X. H.; Zhu, W. H.;
J. Phys. Chem. A2006, 110, 3797.
40. Xu, X. J.; Xiao, H. M.; Ma, X. F.; Ju, X. H.; Int. J. Quantum Chem.2006, 106, 1561.
41. Politzer, P.; Martinez, J.; Murray, J. S.; Concha, M. C.; Toro-Labbé, A.; Mol. Phys.2009, 107, 2095
42. Lide, D. R.; CRC Handbook of Chemistry and Physics; CRC Press LLC: Boca Raton, Florida, USA, 2002.
43. Archibald, T. G.; Gilardi, R.; Baum, K.; George, C.; J. Org. Chem. 1990, 55, 2920.
44. Politzer, P.; Murray, J. S.; Cent. Eur. J. Energetic Mater. 2011,
3, 209.
45. Agrawal, J. P.; Hodgson, R. D.; Organic Chemistry of Explosives; John Wiley & Sons: Chinchester, UK, 2007.
46. Pagoria, P. F.; Lee, J. S.; Mitchell, A. R.; Schmidt, R. D.;
Thermochim. Acta2002, 384, 187.
47. Zhang, C. Y.; Shu, Y. J.; Huang, Y. G.; Zhao, X. D; Dong, H. S;
J. Phys. Chem. B2005, 109, 8978.
Submitted: February 20, 2013