Mecanismos moleculares envolvendo a alfa-sinucleína e a sinfilina-1 na doença de Parkinson
100
0
0
Texto
(2) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. !. Agradecimentos Ao meu orientador, Professor Alexandre Quintas, pela sua disponibilidade, apoio, compreensão e paciência ao longo da realização deste trabalho. Que sem a sua ajuda não seria possível terminar esta etapa. Ao meu querido Pai, porque sem ele nada disto seria possível. Muito obrigado Pai, por tudo. Por tornares tudo possível. Por fazeres com que fosse possível concluir o Grau de Mestre em Ciências Farmacêuticas. Obrigado. A um pai extraordinário. À minha Mãe por todo o apoio. Porque ‘’Quem tem mãe tem tudo’’. Obrigado por seres a melhor mãe que alguma vez poderia ter desejado. Ao Luís Timóteo Ramos, pela paciência infindável, pelo apoio, pela força e por acreditar em mim. Sempre. Espero ter a oportunidade de retribuir todo o apoio. Aos meus amigos, que me acompanharam durante estes anos. Fizeram com que fossem uns anos maravilhosos.. ‘’Souvent il m'arrivait Devant mon chevalet De passer des nuits blanches Retouchant le dessin’’ Charles Aznavour !. !. 2!.
(3) Resumo. Resumo A Doença de Parkinson (DP) é a patologia neurodegenerativa mais prevalente logo a seguir à doença de Alzheimer, afectando 1% dos indivíduos com idades superiores a 60 anos. A DP é caracterizada, clinicamente, por severos sintomas motores incluindo tremor, rigidez muscular, instabilidade postural e bradicinesia. Uma das principais características é a presença de Corpos de Lewy associada a uma perda substancial dos neurónios dopaminérgicos na substância nigra pars compacta. A DP é uma doença multifactorial de etiologia desconhecida, em que tanto os factores genéticos como os ambientais têm uma influência notória. Diversos mecanismos patogénicos podem estar envolvidos na patogénese da DP, incluindo um aumento excessivo dos níveis de stress oxidativo e disfunções a nível da mitocôndria, associado à presença de proteínas misfolding e alterações no sistema ubiquitinaproteassoma assim como no sistema autofagia-lisossoma. A DP permanece incurável, sendo a farmacoterapia com levodopa a mais eficaz no controlo da sintomatologia e considerada a terapêutica de primeira linha. Ainda não é possível abrandar ou parar o processo degenerativo característico da DP através da terapêutica farmacológica disponível. A alfa-Sinucleína, uma pequena proteína expressa no cérebro, é um dos principais componentes dos Corpos de Lewy. A alfa-Sinucleína encontra-se ulfolded no seu estado nativo e pode ter um papel importante a nível da transmissão sináptica e na fisiologia dos neurónios dopaminérgicos. Pensa-se que esta proteína está fortemente relacionada com a patogénese da DP e a toxicidade induzida pela alfa-Sinucleína pode envolver diversos mecanismos, como a agregação ou interacção com outras proteínas e moléculas, incluindo a Sinfilina-1. A Sinfilina-1 é uma proteína citoplasmática que se localiza perto das vesículas sinápticas e colocaliza-se com a alfa-Sinucleína dentro dos Corpos de Lewy. A Sinfilina-1 interage tanto in vitro como in vivo com a alfa-Sinucleína promovendo a sua agregação, o que pode influenciar a formação dos Corpos de Lewy nos neurónios.. Palavras-Chave : alfa-Sinucleína, Sinfilina-1, Doença de Parkinson, Neurodegeneração. 3!.
(4) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. !. Abstract Parkinson’s Diesease is one of the most common neurodegenerative diseases, affecting 1% of individuals over 60 years old. PD is characterized clinically by severe motor symptoms including uncontrollable resting tremor, muscular rigidity, postural instability, and bradykinesia. The pathological hallmarks are the presence of Lewy bodies and massive lost of dopaminergic neuros in the pars compacta of the substantia nigra. PD is a multifactorial disease with unknow etiology, in which both genetic and environmental factors play importante roles. Various pathogenic mechanisms may be involved in pathogenesis of PD including abnormally increased oxidative stress and mitochondrial dysfunction, together with protein misfolding and impairments in the ubiquitin–proteasome and autophagy-lysosomal systems. PD remains a uncurable disease, pharmacotherapy with levodopa represents an effective symptomatic recourse and remains a clinical gold standard treatment for PD, however, any currently available therapies could slow our stop the degenerative process in PD brain. The major component of Lewy body is alpha-synuclein, a small protein which is widely expressed in the brain. Alpha-sinuclein is natively unfolded protein whitch may have an important role in sinaptic transmission and dopaminergic neuron physiology. This protein is thought to be critically implicated in PD pathophysiology and the mechanism by which alpha-sinuclein induces neuronal cell toxicity may involve multiple pathways, such as aggregation or interaction with other proteins and molecules, including synphilin-1. Synphilin-1 represents a cytoplasmatic protein that localizes close to synaptic vesicles and colocalizes with alpha-synuclein into Lewy bodies. Synphilin-1 interacts both in vitro and in vivo with alpha-synuclein promoting its agregation, witch may have implications for Lewy bodies formation in neural cells. Keywords : Alpha-synuclein, Synphilin-1, Parkinson’s Disease, Neurodegeneration. !. 4!.
(5) Índice Geral. Índice Geral Agradecimentos. 2. Resumo. 3. Abstract. 4. Índice Geral. 5. Índice De Figuras. 7. Índice De Tabelas. 8. Abreviaturas. 9. 1. Introdução. 10. 1.1. Doença De Parkinson. 10. 1.1.1. Aspectos Históricos. 10. 1.1.2. Fisiopatologia. 13. 1.1.3. Indutores e Mecanismos da Génese da Doença de Parkinson. 16. 1.1.3.1.. Stress Oxidativo. 16. 1.1.3.2.. Disfunção Mitocondrial. 17. 1.1.3.3.. Disfunção do Sistema Ubiquitina-Proteassoma. 21. 1.1.4. Mecanismos Moleculares. 25. 1.1.4.1.. Factores Ambientais. 25. 1.1.4.2.. Factores Genéticos. 30. 2. Diagnostico e Farmacoterapia na Doença de Parkinson. 36. 2.1. Quadro Clínico e Diagnostico da DP. 36. 2.2. Terapêutica Farmacológica na Doença de Parkinson. 38. 3. Mecanismos Moleculares da Doença de Parkinson Envolvendo a alfa-. 44. Sinucleína e a Sinfilina1 3.1. alfa-Sinucleína. 44. 3.1.1. Estrutura e Localização. 44. 3.1.2. Funções da alfa-Sinucleína. 45. 3.1.3. Envolvimento na doença de Parkinson. 49. 3.2. Sinfilina-1. 60. 3.2.1. Estrutura e Localização. 60. 3.2.2. Funções da Sinfilina-1. 62. 3.2.3. Envolvimento na doença de Parkinson. 63. 5!.
(6) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. ! 3.3. Interacção da alfa-Sinucleína com a Sinfilina-1. 68. 3.4. Agregação Proteica e a Formação de Corpos de Inclusão : Toxicidade ou. 70. Neuroprotecção? 4. Considerações Finais. 75. 5. Referências Bibliográficas. 77. !. 6!.
(7) Índice de Figuras. Índice De Figuras Figura 1 - Anatomia e Fisiologia da DP. 14. Figura 2 - Alterações da Mitocôndria.. 20. Figura 3 - Via da Ubiquitina-Proteassoma. 22. Figura 4 - Mecanismos Neurodegenerativos. 24. Figura 5 - Representação Esquemática do Metabolismo Do MPTP. 26. Figura 6 - Representação Esquemática do Metabolismo Do MPP+.. 27. Figura 7 - Representação Esquemática da Estrutura da α-Syn. 45. Figura 8 - Representação Esquemática do Processo de Agregação da α-Syn. 51. Figura 9 - Proposta de Modelo da Toxicidade da α-Syn. 57. Figura 10 - Visão Esquemática da Sinfilina-1. 62. Figura 11- Representação Esquemática da Sinfilina-1 e da Sinfilina-1A. 67. Figura 12 - Representação Esquemática da Interacção entre a α-Syn e a Sinfilina-1. 68. Figura 13 - Representação Esquemática da Formação de Corpos de Inclusão em Modelos Celulares da DP. 72. 7!.
(8) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. !. Índice De Tabelas Tabela 1 - Resumo dos Genes Envolvidos na DP. 35!. Tabela 2 - Critérios de Diagnóstico da DP. 37!. Tabela 3 - Fármacos Agonistas dos Receptores de Dopamina. 41!. !. 8!.
(9) Abreviaturas. Abreviaturas ! AADC - Descarboxilase Dos Aminoácidos L-Aromáticos Ach- Acetilcolina (do inglês Acetylcholine) BHE- Barreira Hematoencefálica cI- Complexo I CL- Corpos de Lewy CMA- Autofagia Mediada por Chaperonas (do inglês Chaperone-mediated autophagy COMT%&%Catecol-O-Metiltransferase% CTE – Cadeia Transportadora de Electrões DAT- Transportadores de Dopamina (do inglês dopamine active transporter) DP- Doença de Parkinson GPi- Globo Pálido Interno GSH%&%!Glutationa!(do!inglês!Glutathione)! GSK3B- Glicogénio Quinase-3-. (do inglês Glycogen synthase kinase 3 beta). L&dopa%&%Levodopa! NOS- Óxido Nítrico Sintetase (do inglês nitric oxide synthase) PP2A%& Proteína Fosfatase 2A% Pu- Putamên ROS- Espécies Reactivas de Oxigénio (do inglês Reactive Oxygen Species) SNC- Sistema Nervoso Central SNpc- Substância nigra pars compacta SOD- Superóxido Dismutase SUP- Sistema Ubiquitina-Proteassoma Th- Tálamo (do inglês Thalamus) VMAT- Transportadores Vesiculares de Monoamina (do inglês vesicular monoamine transporter) WT%–%Wild-Type1 α-Syn- alfa-Sinucleína (do inglês Alpha-Synuclein). 9!.
(10) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. !. 1. Introdução 1.1 Doença De Parkinson A Doença de Parkinson (DP) é uma doença neurológica crónica e progressiva associada a uma perda substancial de neurónios dopaminérgicos da substancia nigra pars compacta (SNpc) e outras alterações complexas que conduzem ao aparecimento de diversos sintomas motores e não-motores (Perfeito, Cunha-Oliveira, & Rego, 2012). É a segunda doença neurodegenerativa mais comum, apresentando um risco de 1% após os 60 anos e que aumenta até 4% após os 80 anos (Dexter & Jenner, 2013; Lau & Breteler, 2006). Tanto a incidência como a prevalência da doença aumentam com a idade, contudo, são ligeiramente superiores no sexo masculino (F. J. S. Lee & Liu, 2008). Na Europa, a prevalência varia desde 65.6/100.000 a 12500/100.00 sendo a incidência anual de 5/100 000 a 346/100 000 (Pereira, 2011; von Campenhausen et al., 2005). ! 1.1.1. Aspectos Históricos. Apesar de já ser conhecida previamente na antiga Índia pelo nome de ‘’Kampavata’’, a Doença de Parkinson foi descrita pela primeira vez numa monografia, em 1817, por James Parkinson no seu estudo ‘’An Essay on the Shaking Palsy’’ (Corti, Lesage, & Brice, 2011; Teive, 1998). James Parkinson intitula a efemeridade como ‘’Paralisia Agitante’’ e caracteriza a doença pela presença de movimentos trémulos involuntários, com diminuição da força muscular, com tendência para a inclinação do tronco para a frente e com alteração da marcha, sendo os sentidos e o intelecto não afectados (Parkinson, 2002; Teive, 1998). ‘’Involuntary tremulous motion, with lessened muscular power, in parts not in action and even when supported; with a propensity to bend the trunk forwards, and to pass from a walking to a running pace: the senses and intellects being uninjured.’’(Parkinson, 1817, 2002) No entanto, foi Jean-Martin Charcot, considerado o pai da Neurologia, que teve um contributo extraordinário na definição clínica da Doença de Parkinson. Primariamente, !. 10!.
(11) Introdução. foi Jean-Martin Charcot quem sugeriu a mudança de nome para Doença de Parkinson em homenagem à descrição clássica de James Parkinson. Por outro lado, Charcot definiu a presença dos sinais mais comuns da doença: tremor, bradicinesia, rigidez e dificuldade do equilíbrio. Apresentando os critérios para o diagnóstico diferencial da doença. Por fim, em 1877, Charcot prescreve a primeira terapêutica para a DP, a hioscinamida, um percussor dos alcaloides da beladona com propriedades anticolinérgicas (Teive, 1998). Em 1888, Gowers publica o livro ‘’Manual of Diseases of the Nervous System’’ onde descreve a sua experiencia com 80 pacientes com a Doença de Parkinson. Gowers nota que existe uma predominância masculina na incidência da doença, com idades compreendidas entre 50-60 anos e observou que cerca de 15% dos seus pacientes tem uma forte história familiar, referindo pela primeira vez a vertente genética da Doença de Parkinson (C. Goetz, Chmura, & Lanska, 2001; J.William Langston, 2002; Pearce, 1989). Brissaud, em 1895, comenta ‘’a lesion of the locus niger could very well be the anatomical basis of Parkinsons disease’’, referindo o envolvimento do mesencéfalo na DP (C. Goetz et al., 2001; Pearce, 1989). Contudo foi Trétiakoff, em 1919, quem confirmou a hipótese de Brissaud ao estudar a substância nigra do cérebro de 54 pacientes dos quais 9 sofriam da Doença de Parkinson. Trétiakoff notou que em todos os casos com DP havia uma notória diminuição da substância negra mesencefálica (Parent & Parent, 2010). Trétiakoff também encontrou, no cérebro dos pacientes com DP, umas inclusões concêntricas no citoplasma dos neurónios da substância negra, aos quais denominou de Corpos de Lewy (CL) em homenagem a Friedrich Lewy que descreveu estas inclusões plasmáticas pela primeira vez em 1913 (Parent & Parent, 2010; Pearce, 1989). Por volta de 1953, Greenfield e Bosanquet forneceram a mais completa análise patológica da DP e das lesões a nível cerebral (C. Goetz et al., 2001; C. G. Goetz, 2011). Em 1967, Hoehn e Yahr, descrevem a morbilidade e evolução clínica da doença, desenvolvendo a escala de Hoehn e Yahr que indica o estado geral do paciente (C. G. Goetz, 2011). A escala de HY, originalmente, dividia-se em 5 estágios : nos estágios I, II e III considerava-se que o doente apresentava uma incapacidade de leve a moderada, enquanto que nos estágios IV e V a incapacidade do doente era classificada como mais grave (Goulart & Xavier, 2005).. 11!.
(12) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. ! O estudo farmacológico e bioquímico da DP começou na época de 1960’s com Ehringer e Hornykiewicz a documentarem a perda de dopamina no tecido estriado do cérebro em pacientes com Doença de Parkinson. Pouco tempo depois, Barbeau testa o tratamento com Levodopa (L-dopa) per os e Birkmater e Hornykiewicz testam, independentemente, o uso de L-Dopa por via intravenosa (C. G. Goetz, 2011).. !. 12!.
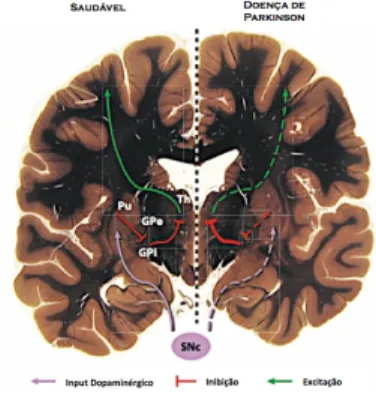
(13) Introdução. 1.1.2 Fisiopatologia A Doença de Parkinson é caracterizada por uma diminuição progressiva dos neurónios da substância nigra pars compacta com a presença de depósitos de proteínas no citoplasma dos neurónios (CL). Aquando da morte do paciente, a SNpc apresenta uma perda de cerca de 50-70% comparando com a mesma região em indivíduos saudáveis (Davie, 2008; Rodrigues & Campos, 2006). Na fase inicial da doença, estágio 1 e 2, ocorrem alterações a nível do bulbo olfactivo e da medula oblonga, neste estágio inicial o paciente apresenta-se num estado présintomático. À medida que a doença progride, a SNpc e outras regiões do cérebro como o mesencéfalo, o prosencéfalo basal e o neocortex são afectadas, atingindo-se os estágios 3 e 4 da DP (Davie, 2008). A perda de células dopaminérgicas conduz directamente a uma diminuição dos níveis de dopamina na SNpc (Khatri & Chaudhry, 2009). A diminuição de dopamina leva a uma activação reduzida dos seus receptores provocando uma redução da inibição da via indirecta e uma diminuição da excitação da via directa. A inibição reduzida da via indirecta provoca uma inibição excessiva do globus pallidus pars externa, a desinibição dos gânglios da base e um aumento da excitação dos neurónios do globus pallidus pars interna e da substância nigra pars compacta, ao passo que a diminuição da activação da via directa causa uma diminuição da sua influência inibitória sobre o globus pallidum pars interna e sobre a substância nigra pars compacta (Bartels & Leenders, 2009; Obeso & Rodriguez-Oroz, 2000). Resultando numa actividade excessiva do corpo estriado que provoca uma aferência contínua de sinais excitatórios para o controlo motor córtico-espinhal, o que resulta num desequilíbrio entre o sistema excitatório e inibitório comprometendo o controlo motor, conduzindo ao aparecimento das características da DP (Bartels & Leenders, 2009).. 13!.
(14) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. !. Figura 1- Anatomia E Fisiologia Da DP. Esquema simplificado dos circuitos neuronais envolvendo o núcleo da base, o tálamo (Th) e o córtex e as alterações sofridas por estas estruturas na DP. Apenas está representada a via directa que em, condições normais, (esquerda) facilita os movimentos, contudo, na DP, encontra-se atenuada (direita). A SNpc fornece os estímulos dopaminérgicos (Input Dopaminérgico) ao putamên (Pu), o que leva á excitação da via directa. O Pu inibe (vermelho) o globo pálido interno (GPi) que consequentemente inibe o Th. O Th envia estímulos excitatórios (verde) para o córtex motor. Na DP, a degeneração da SNpc conduz a uma inibição dos estímulos tálamo-corticais. A via indirecta (não representada) é inibida pelos estímulos dopaminérgicos e, em condições fisiológicas, reprime os movimentos, contudo, a actividade desta via encontra-se aumentada na DP. (Adaptado de Shulman, De Jager, & Feany, 2011). Como já foi referido anteriormente, a presença de CL, inclusões citoplasmáticas eosinofílicas com núcleos densos rodeados por filamentos radiados, é característico da DP. Os CL são formados por diversas proteínas como a ubiquitina e a alfa-Sinucleína (α-Syn) entre outras, estas podem estar homogeneamente distribuídas ou haver uma concentração de ubiquitina no centro dos CL e de α-Syn na periferia o que é consistente com a hipótese de formação de depósitos pela α-Syn (Tatsch, Nitrini, & Neto, 2002). Os CL são depositados em diferentes partes dos cérebro consoante a progressão da DP. No estágio inicial da DP as lesões aparecem no núcleo dorsal motor IX/X e na zona reticular intermediaria, havendo uma extensão das lesões à medida que a doença se desenvolve acabando por atingir, numa fase mais avançada da DP (estágios 4, 5 e 6) o neocortex. O estágio 2 da DP acrescenta lesões a nível do núcleo reticular giantocelular e do complexo coeruleus-subcoeruleus. As lesões na substância nigra pars compacta relacionam-se com a DP no estágio 3 assim como outras lesões a nível do mesencéfalo. No estágio 4 as lesões atingem o prosencéfalo, o mesocortex temporal e o alocórtex. O. !. 14!.
(15) Introdução. neocortex só é afectado no estágio 5 juntamente com o neocortex pré-frontal. Por fim, no estágio 6 da DP observam-se lesões a nível do neocortex e nas áreas pré-motoras associadas as funções sensoriais (Leong, Cappai, Barnham, & Pham, 2009). Apesar de o papel destas inclusões na patogénese da DP não estar totalmente esclarecido, a descoberta da α-Syn como um dos compostos principais dos filamentos radiados nos CL veio alterar o ponto de vista sobre a formação e o papel dos destes nos processos neurodegenerativos. Levando à formulação de novas prespectivas fisiopatologicas sobre a progressão da DP e a sua classificação como uma Sinucleinopatia (Dexter & Jenner, 2013).. 15!.
(16) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. ! 1.1.3. Indutores e mecanismos da génese da Doença de Parkinson. 1.1.3.1 Stress Oxidativo O stress oxidativo resulta da eliminação insuficiente das espécies reactivas de oxigénio (ROS) geradas por diversas reacções bioquímicas. Em condições fisiologicas são eliminadas pelos sistemas antioxidantes intracelulares que podem estar afectados em condições patogénicas ou com o envelhecimento natural (Gómez-Chavarín, RoldanRoldan, Morales-Espinosa, Pérez-Soto, & Torner-Aguilar, 2012). Os neurónios dopaminérgicos estão especialmente expostos ao stress oxidativo devido ao metabolismo da dopamina que origina diversas moléculas que actuam como toxinas endógenas se não forem eliminadas correctamente. A dopamina possui duas vias de oxidação : (1) Pode sofrer auto-oxidação ao reagir com o oxigénio a pH neutro e originando espécies dopamina-quinona tóxicas, radicais superóxidos e peroxido de oxigénio; ou (2) Ser metabolizada pela MAO em DOPAC (um metabolito não tóxico) e em peroxido de oxigénio (Levy, Malagelada, & Greene, 2009) . Apesar do superóxido não ser uma molécula altamente reactiva, pode ser convertida em peroxido de oxigénio pela oxigénio dismutase ou em radicais peroxinitrito na presença de óxido nítrico. O peroxido de oxigénio por sua vez pode ser convertido em radicais hidroxilo tóxicos através da Reacção de Fenton que ocorre em presença de elevados níveis de ferro, cujas concentrações são bastante elevadas na SNpc (Teive & Menezes, 2003). As ROS provocam alterações funcionais a nível das proteínas, lípidos e DNA (Levy et al., 2009). Os danos a nível dos lípidos, por sua vez, levam a um aumento da permeabilidade da membrana a iões, tal como o cálcio que podem vir a promover a exotoxicidade (Lotharius & Brundin, 2002). Como a dopamina citoplasmática pode formar espécies reactivas de oxigénio muito rapidamente, tem de ser sintetizada ou transportada do espaço extracelular para o interior da célula e armazenada no interior das vesículas sinápticas (Jenner, 2003). Assim, são várias as circunstâncias que vão levar a um aumento dos níveis de stress oxidativo e consequentes danos celulares a nível do SNC : (a) O aumento dos níveis de dopamina; (b) Deficiência em glutationa (GSH) que provoca a diminuição da capacidade do cérebro para eliminar o peroxido de oxigénio; (c) Um aumento dos depósitos de ferro; (d) Danos no DNA; (e) Peroxidação dos lípidos (C. Olanow & Tatton, 1999).. !. 16!.
(17) Introdução. Na DP todas as células da SNpc aparentam estar sob um elevado estado de stress oxidativo (Gómez-Chavarín et al., 2012). De facto, diversos estudos post-mortem em cérebros de indivíduos com DP revelaram elevados níveis de ferro, GSH diminuída e danos oxidativos nos lípidos, proteínas e DNA (C. Olanow & Tatton, 1999) Outra evidência de ocorrência de stress oxidativo na DP, surge aquando dos estudos sobre o parkinsonismo induzido pelo MPTP, que através da redistribuição de dopamina no citoplasma, promove o aumento do stress oxidativo (Di Monte, 2001; Lotharius & Brundin, 2002). Também os pesticidas rotenona e paraquato, demonstraram alterar os níveis de dopamina no citoplasma e consequente aumento do stress oxidativo (GómezChavarín et al., 2012). Estes factos sugerem que os factores ambientais podem, também, estar envolvidos no aumento do stress oxidativo. Contudo, a origem dos elevados níveis stress oxidativo a que o SNC esta exposto na DP continua a ser bastante controversa. No entanto, é claro que a acumulação de ferro no SNC leva a alterações na actividade dos canais de cálcio, altera a proteólise e provoca alterações a nível da agregação da α-Syn. Todas estas ocorrências enfatizam a natureza diversa das causas de morte celular na DP e também o contributo do stress oxidativo nos mecanismos neurodegenerativos (Dexter & Jenner, 2013).. 1.1.3.2 Disfunção Mitocondrial A mitocôndria é um organelo citoplasmático essencial para diversos mecanismos celulares incluindo a produção de ATP, regulação de ROS, homeostase do cálcio e o controlo da apoptose (Fujita et al., 2013). Na sua organização estrutural, apresenta duas membranas, interna e externa, envolvendo uma matriz. Na membrana mitocondrial interna está localizada a cadeia transportadora de electrões (CTE), constituída por cinco complexos I-V, onde ocorre a produção de energia através do fluxo de electrões e consequente fosforilação oxidativa (fonte principal de energia das células eucarióticas) (Nasseh & Tengan, 2001). Devido ao processo de fosforilação, as mitocôndrias são a principal fonte celular de radicais livres, pois este mecanismo ocorrente na CTE promove a formação de espécies extremamente reactivas, nomeadamente os radicais superóxidos gerados no complexo I (cI). A inibição deste complexo provoca a depleção de ATP e danifica todos os mecanismos celulares dependentes de ATP, promovendo a formação de radicais livres e consequentemente, o aumento do stress oxidativo (Júnior & Aguiar, 2007). Estas. 17!.
(18) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. ! espécies reactivas de oxigénio, geradas pela CTE, concomitantemente com os danos nas membranas celulares, são a causa mais frequente de mutações a nível do DNA mitocondrial (mtDNA) e provocam também o aumento de proteínas misfolded, contribuindo para danos a nível do Sistema Ubiquitina-Proteassoma (SUP) (Buneeva & Medvedev, 2011; Dauer & Przedborski, 2003; Levy et al., 2009) A disfunção mitocondrial é, assim, caracterizada por (a) Alterações na CTE resultando num aumento das espécies reactivas de oxigénio e consequente aumento do stress oxidativo (Hastings, 2009); (b) Produção insuficiente de ATP (Levy et al., 2009); (c) Activação das caspases (proteínas essenciais na regulação da apoptose) (Levy et al., 2009). Na DP, diversas evidências sugerem uma disfunção no complexo I, havendo uma redução da actividade deste complexo na SNpc (Levy et al., 2009). O recurso a modelos utilizando DNA mitocondrial de indivíduos com PD esporádica demonstrou uma reduzida actividade do complexo I, implicando uma procedência de alterações no mtDNA, contudo, não foram demonstradas alterações consistentes no mtDNA (Dexter & Jenner, 2013; Gu, Cooper, Taanman, & Schapira, 1998). Uma outra evidência da disfunção mitocondrial na DP está relacionada com o MPTP, pois após a administração, este composto vai interferir com a CTE inibindo o complexo I com uma consequente diminuição dos níveis de ATP e aumento do stress oxidativo (Dauer & Przedborski, 2003; Koller, Vetere-Overfield, & Gray, 1990). O mecanismo deste composto será explicado no próximo capítulo. Um outro composto tóxico relacionado com a disfunção mitocondrial é a rotenona, um pesticida que também interfere com a CTE (Levy et al., 2009). A manutenção da homeostase mitocondrial através da mitofagia envolve diversos factores que controlam diversos processos abrangendo desde a fusão e fissão da mitocôndria até à mobilidade. Estes processos aparentam estar fortemente relacionados com as proteínas envolvidas nas formas esporádicas e familiares da DP (Fujita et al., 2013). Diversas mutações relacionadas com a DP foram associadas com alterações da função da mitocôndria. Estas mutações podem levar a uma alteração da localização das proteínas na mitocôndria, anomalias estruturais e funcionais e modificações na CTE (Dexter & Jenner, 2013). Deste modo, são várias as proteínas associadas a DP familiar que estão relacionadas com a mitocôndria, com destaque para a PINK1 e Parkina. A actividade quinase da PINK1, tendo como alvos as proteínas Parkina, HTR2 e TRAP1, controla o turnover !. 18!.
(19) Introdução. mitocondrial e tem funções de protecção contra o stress oxidativo e, por outro lado, a mitofagia é impulsionada pela translocação da Parkina do citosol para a mitocôndria mediada pela PINK1 (Chan et al., 2011; Matsuda, Sato, & Shiba, 2010; Plun-Favreau, Klupsch, & Moisoi, 2007; Pridgeon, Olzmann, Chin, & Li, 2007). Também a mitofagia e o controlo transcricional da biogénese da mitocôndria estão dependentes da actividade da ligase de ubiquitina E3 da Parkina. A Parkina é uma proteína que esta associada à membrana mitocondrial externa e actua atrasando o swelling mitocondrial e consequente libertação do citocromo c e activação da caspase 3, a perda desta função em pacientes com mutações no gene Parkina parece contribuir para a degeneração dos neurónios dopaminérgicos (Darios, Corti, & Lücking, 2003). Ratinhos knockout para a Parkina e PINK1 demonstraram uma reduzida função respiratória mitocondrial concomitantemente com uma morfologia não alterada da mitocôndria e sem sinais de neurodegeneração, contudo, ambos os animais mutados expressaram elevados níveis de stress oxidativo (Levy et al., 2009). Os genes associados à DP familiar parecem estar relacionados, também, com a produção de ROS pela mitocôndria. A deficiência em PINK1 altera a homeostase do cálcio e os mecanismos de respiração mitocondrial (Gandhi et al., 2009; Yao et al., 2011). Consequentemente, a diminuição dos níveis de cálcio e o aumento de ROS podem alterar a permeabilidade da membrana exterior da mitocôndria conduzindo à degeneração celular (Gandhi et al., 2009). Estudos sugerem que as funções da proteína DJ-1 estão associadas ao complexo PINK1-Parkina na manutenção da função mitocondrial na presença de stress oxidativo (Thomas et al., 2011). A proteína DJ-1, no seu estado oxidado, parece ter um efeito protector contra o stress oxidativo enquanto a PINK1 localiza-se na matriz mitocondrial e desempenha funções de protecção contra a apoptose, efeito que se encontra reduzido nas mutações associadas à DP (Kim et al., 2005; Petit et al., 2005; Silvestri et al., 2005). Também a α-Sinucleína parece estar relacionada com a mitocôndria, num estudo realizado em ratinhos transgénicos, o aumento da expressão de α-Syn comprometeu a função mitocondrial, aumentado o stress oxidativo e a toxicidade do MPTP (Song, Shults, Sisk, Rockenstein, & Masliah, 2004).. 19!.
(20) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. !. Figura 2 - Alterações Na Mitocôndria. As alterações a nível da mitocôndria são observadas em diversas doenças neurodegenerativas. Inibidores do complexo I, como o MPTP e a rotenona, causam sintomas semelhantes aos da DP. Para além disso, proteína envolvidas nas formas familiares da DP, como a LRRK2, a α-Syn, a Parkina, a DJ-1 e a PINK-1 encontram-se associadas à membrana externa (ME) da mitocôndria e estão envolvidas na produção de ROS. A HTRA, uma outra proteína que se encontra mutada na DP familiar, localiza-se no espaço intramembranar (EIM) e pode estar envolvida no processamento proteólico das proteínas mitocondriais. Mutações nos mecanismos de fissão e fusão mitocondrial podem causar doenças neurodegenerativas e, de facto, as proteínas PINK-1 e Parkina parecem estar envolvidas na fissão mitocondrial, para além do seu papel nas funções normais da mitocôndria. (Adaptado de Knott, Perkins, Schwarzenbacher, & Bossy-Wetzel, 2008). ! ! ! ! ! ! ! ! ! !. 20!.
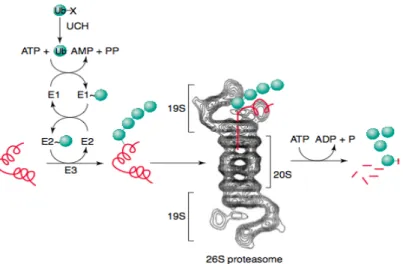
(21) Introdução. 1.1.3.3 Disfunção do Sistema Ubiquitina-Proteassoma Em células como os neurónios, os sistemas de degradação de proteínas danificadas ou misfolded assumem um papel de elevada importância de forma a manter a homeostase. A disfunção destes sistemas de controlo de qualidade das proteínas, como o SUP, especialmente na sinapse podem levar a acumulação de proteínas, como a αSyn, que por sua vez interferem com as funções sinápticas, o que proporciona a formação de agregados tóxicos ou a alterações perniciosas no metabolismo energético (Fujita et al., 2013). O SUP é essencial para a degradação de proteínas danificadas nas células eucarióticas (Ross & Pickart, 2004). O mecanismo de degradação de proteínas pelo SUP é um processo que envolve diversas reacções mediadas por enzimas e através de moléculas de ubiquitina (Mcnaught, Olanow, Halliwell, Isacson, & Jenner, 2001). A proteína danificada é identificada e sinalizada por moléculas de ubiquitina, que se ligam de forma covalente à proteína, para posterior degradação (Betarbet, Sherer, & Greenamyre, 2005). Primariamente a ubiquitina é activada, por um mecanismo dependente de ATP, pela E1 (enzima activadora da ubiquitina), seguidamente a proteína alvo liga-se à enzima E2 (enzimas conjugadoras de ubiquitina), sendo uma reacção catalisada pela enzima E3 (ligase de ubiquitina-proteína), formando um conjugado proteína-ubiquitina que é posteriormente reconhecido e degradado pelo proteassoma S26 (Figura 3) (Mcnaught et al., 2001; Pickart & Eddins, 2004).. 21!.
(22) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. !. ! Figura 3 - Via da Ubiquitina-Proteassoma. Na sequência do processo pela enzima UCH, a ubiquitina é conjugada com um substrato específico (vermelho) numa cadeia poliubiquitinada. A cadeia poliubiquitinada direcciona o substrato para o proteassoma S26, constituído por um complexo proteolítico 20S e dois complexos reguladores 19S. A cadeia polipeptidica é desenrolada e mobilizada para dentro do complexo 20S. O substrato é hidrolisado em pequenos péptidos enquanto a ubiquitina é reciclada. As enzimas E1, E2 e E3 são respectivamente activadoras da ubiquitina, conjugadoras de ubiquitina e ligases da ubiquitina. (Adaptado de Ross & Pickart, 2004). A presença de diversas proteínas nos CL, incluindo a α-Syn, levou à formulação da hipótese de que o catabolismo das proteínas danificadas ou mutadas poderia estar alterado, provocando a agregação celular e a morte dos neurónios. O SUP passou, então, a ser considerado como uma das causas prováveis da fisiopatologia da DP (Dexter & Jenner, 2013; Onyango, 2008). O envolvimento do SUP na DP é, também, suportado pela descoberta de mutações nas proteínas Parkina e UCH-L1, pois estas proteínas funcionam, respectivamente, como ligases no complexo proteína-ubiquitina e possuem funções de reciclagem da ubiquitina (Dexter & Jenner, 2013). A primeira indicação do envolvimento do SUP na DP foi a detecção de ubiquitina e subunidades proteassomais nos CL sugerindo uma disfunção do SUP (Beyer, DomingoSàbat, & Ariza, 2009; Levy et al., 2009). A detecção de uma diminuição da actividade do proteassoma na SNpc em indivíduos com DP suporta, também o envolvimento do SUP na DP (Beyer et al., 2009). A actividade proteassomal também diminui com o envelhecimento natural, especialmente na SNpc, o que pode contribuir para a vulnerabilidade selectiva da SNpc (Levy et al., 2009).. !. 22!.
(23) Introdução. A identificação da mutação no gene Parkina associada à DP suporta o envolvimento do SUP na DP. A proteína Parkina é uma ligase E3 e as mutações nesta proteína, relacionadas com a DP, provocam uma diminuição da actividade desta. Por outro lado, a Parkina também foi identificada nos CL e os níveis de Parkina na forma nitrosilada estão aumentados na SNpc na DP, o que leva a uma diminuição da actividade desta. In vitro, foram identificados diversos substratos desta proteína, incluindo a Sinfilina-1 e a α-Syn, cujas expressões em excesso provoca a morte dos neurónios dopaminérgicos. Estes mesmos substratos encontram-se em elevadas concentrações na SNpc e/ou nos CL nas autopsias de indivíduos com DP esporádica (Levy et al., 2009; Mcnaught et al., 2001). A olígomerização da α-Syn e a sobre-expressão desta parecem contribuir para a disfunção do SUP (Onyango, 2008). A sobre-expressão de α-Syn em células PC12 provocou uma inibição da actividade proteassomal, possivelmente pela ligação dos olígomeros da α-Syn ao próprio proteassoma (Stefanis, Larsen, Rideout, Sulzer, & Greene, 2001). Por outro lado, a inibição do SUP contribui para a formação de agregados e acumulação da α-Syn nas inclusões citoplasmáticas (Levy et al., 2009). O estudo do proteassoma 26S, na DP, revelou alterações na actividade catalítica e na sua composição na SNpc estando possivelmente associado ao comprometimento da degradação da α-Syn (Dexter & Jenner, 2013). A inibição da mitocôndria leva a uma diminuição dos níveis de ATP intracelulares afectando a degradação proteassomal, um processo dependente de ATP sugerindo que a interacção entre o SUP e a mitocôndria pode promover os processos neurodegenerativos nos neurónios dopaminérgicos (Onyango, 2008).. 23!.
(24) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. !. ! Figura 4 - Mecanismos Neurodegenerativos - Os factores de risco conhecidos da DP incluem os factores ambientais (verde), factores genéticos (roxo) e factores endógenos (azul). A contribuição destes factores levam a modificações oxidativas, disfunções da mitocôndria e alterações na degradação de proteínas que, em conjunto, provocam uma série de eventos moleculares relacionados entre si que estão subjacentes à neurodegeneração. As interacções entre estes mecanismos devem-se a diversos motivos: (1) Perturbações na respiração mitocondrial gera espécies reactivas de oxigénio; (2) A sobre-expressão de SOD têm um efeito protector relativamente a toxinas da mitocôndria; (3) A deficiência ou inibição da enzima óxido nítrico sintetase (NOS) atenua a toxicidade do MPTP, do paraquato e da rotenona; (4) A inibição dos sistemas de degradação leva a uma sensibilidade aumentada ao stress oxidativo; (5) A degradação, ao estar comprometida, provoca uma acumulação de substratos aumentando a probabilidade de modificações oxidativas; (6) A excessiva produção de espécies reactivas de oxigénio e nitrogénio modifica as proteínas levando à sua inactivação, crosslinking e agregação; (7) A α-Syn modificada pela dopamina oxidada impede a autofagia mediada por chaperonas (CMA); (8) Alterações oxidativas levam a modificações na membrada das proteínas e do lisossoma; (9) O SUP e a CMA não estão aptos para remover e reparar as proteínas danificadas; (10) As modificações das subunidades do proteassomal, devido à oxidação, inibem o SUP; (11) A macroautofagia é o principal mecanismo de degradação da mitocôndria danificada; (12) A inibição do proteassomal aumenta a formação de espécies reactivas na mitocôndria o que compromete a actividade do complexo I e II. (Adaptado de Malkus, Tsika, & Ischiropoulos, 2009). !. 24!.
(25) Introdução. 1.1.4. Mecanismos Moleculares. Apesar dos esforços para descobrir a etiologia da DP, esta ainda permanece por desvendar. Embora tenham sido relatados casos de DP relacionados com mutações genéticas, a maioria das incidências é esporádica na natureza. Pensa-se que a Doença de Parkinson Idiopática é originada através da associação entre diversos factores de risco como os factores genéticos, ambientais e o envelhecimento natutal (Chinta et al., 2013). 1.1.4.1 Factores Ambientais Diversos estudos neuropatológicos e em modelos animais demonstram que as neurotoxinas ambientais podem determinar um dano progressivo na SNpc muito antes do aparecimentos dos sintomas parkinsonianos. A DP, assim como outras doenças neurológicas relacionadas com a idade, pode ser determinada por exposição a toxinas ambientais desde a gravidez até a vida adulta. No entanto, os estudos epidemiológicos mais recentes têm-se direccionado para os factores de risco ambientais presentes na vida adulta (Logroscino, 2005). Uma das evidências mais marcantes, relativamente aos factores ambientais envolvidos na DP, surgiu aquando da observação que o MPTP provocou o aparecimento de sintomas similares aos característicos da DP, em indivíduos expostos acidentalmente (J W Langston, Ballard, Tetrud, & Irwin, 1983). Diversos estudos epidemiológicos têm sugerido que a exposição a pesticidas, e outras toxinas ambientais, está associado ao desenvolvimento da DP. Através da capacidade destes compostos de interferir com os processos mitocondriais, interromper o metabolismo da dopamina e promover a formação de espécies reactivas de oxigénio podem iniciar uma cascata de eventos pejorativos com capacidade de levar à neurodegeneração observada na DP (Malkus et al., 2009; C. Olanow & Tatton, 1999) MPTP A descoberta, em 1983, que a administração intravenosa de MPTP leva ao desenvolvimento de sintomas típicos da DP e a descoberta posterior que este induz danos nas células dopaminérgicas da SNpc levou à suposição que a exposição a certos. 25!.
(26) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. ! factores ambientais pode determinar um aumento do risco de vir a desenvolver a DP (Lau & Breteler, 2006). O 1-metil-4-fenil-1,2,3,6-tetrahidropiridina (MPTP) é um produto intermediário da síntese de MPPP, um análogo da mepiridina (J. Langston, Ballard, Tetrud, & Irwin, 1983). Em 1983, esta toxina foi administrada (acidentalmente) por um grupo de toxicodependentes (não relacionados entre si) que vieram a desenvolver, irreversivelmente, sintomas similares aos da Doença de Parkinson, tais como: bradicinesia, tremor, rigidez e instabilidade postural (C. Goetz et al., 2001; J. Langston et al., 1983). Esta pró-toxina (MPTP) é um composto lipossóluvel capaz de atravessar a barreira hematoencefálica (BHE). Após atravessar a BHE, o MPTP é convertido em MPP+ num processo bifásico. Primariamente, nos neurónios serotoninérgicos e na glia, é metabolizado pela iMAO-B originando o composto intermediário MPTP+, de seguida, através de um mecanismo desconhecido forma-se o MPP+, o composto activo, que por sua vez é posteriormente libertado no espaço extracelular (Przedborski & Vila, 2001) .. Figura% 5% &% Representação% esquemática% do% metabolismo% do% MPTP.! Após! administração! sistémica,!o!MPTP!atravessa!a!BHE.!Uma!vez!no!cérebro,!é!convertido!em!MPDP+!pela!MAOTB!e! depois! em! MPP+! através! de! um! mecanismo,! para! já,! desconhecido.! Seguidamente,! o! MPP+! é! libertado! no! espaço! extracelular! e! entra! nos! neurónios! dopaminérgicos! através! dos! DAT! (Adaptado!de!Dauer!&!Przedborski,!2003)!!. !. 26!.
(27) Introdução. O MPP+ entra para os neurónios dopaminérgicos através dos transportadores da dopamina (DAT) para os quais possui elevada afinidade. Uma vez dentro destes, o MPP+ pode seguir três vias: (1) Ligar-se aos transportadores vesiculares de monoamina (VMAT), e acumula-se nas vesículas sinaptossomais; (2) Inserir-se na matriz mitocondrial; ou (3) Permanecer no citosol e interagir com enzimas citosólicas (Dauer & Przedborski, 2003; Przedborski & Vila, 2001).. ! Figura 6 - Representação esquemática do metabolismo do MPP+. Uma vez dentro dos neurónios dopaminérgicos, o MPP+ pode seguir 3 vias: (1) Inserir-se na matriz mitocondrial (tóxico); (2) Permanecer no citosol e interagir com enzimas citosólicas (tóxico); (3) ligar-se aos VMAT, e acumulase nas vesículas sinaptossomais (protector). Uma vez na mitocôndria, o MPP+ bloqueia o cI da CTE. (Adaptado de Dauer & Przedborski, 2003). A acumulação do MPP+ dentro das vesículas parece exercer um efeito neuroprotector, impedindo o efeito citotóxico, pois evita a interacção deste metabolito com a matriz mitocondrial (Koller et al., 1990). Uma vez inserido na matriz mitocondrial, o MPP+ vai interferir com a CTE, inibindo o cI originado uma diminuição da fosforilação oxidativa. A inibição desta via provoca uma diminuição dos níveis de ATP encefálicos o que provoca a morte celular (Dauer & Przedborski, 2003). Outro efeito da inibição do cI será um aumento do. 27!.
(28) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. ! stress oxidativo proveniente da formação de ROS, nomeadamente radicais superóxido, o que também vai contribuir para a morte celular (Koller et al., 1990). Paraquato e outros Pesticidas Após a descoberta de uma possível relação entre o MPTP e a DP, foram realizados vários estudos com o intuito de examinar a associação entre a exposição a factores ambientais, tais como exposição a herbicidas e pesticidas, actividades agrícolas, vida rural e o consumo de água provenientes de poços, com o risco acrescido de vir a adquirir a DP (Lau & Breteler, 2006) . Apesar da existência de divergências entre os resultados, possivelmente devido a diferenças na metodologia aplicada e na amostra populacional, existe um padrão geral. Este padrão sugere que o risco de vir a adquirir a DP encontra-se acrescido em ambientes rurais, nomeadamente em agricultores, possivelmente devido à maior exposição a herbicidas e pesticidas (Koller et al., 1990). O herbicida paraquato é uma toxina com efeitos pejorativos nos neurónios. O paraquato foi estudado em modelos animais, mais concretamente em ratinhos, induzindo uma redução da actividade motora e morte celular, especialmente nos neurónios dopaminérgicos da SNpc (McCormack et al., 2002). Adicionalmente, a administração sistémica deste composto resulta num aumento da expressão da α-Syn e consequente formação de agregados, similar às modificações provocadas após a administração de MPTP (Manning-Bog et al., 2002; Vila et al., 2001). A sobreexpressão da enzima superóxido dismutase (SOD), em ratinhos e em culturas celulares, relevou um efeito protector em relação à toxicidade provocada pelo paraquato, o que evidência o papel do stress oxidativo na neurodegeneração (Clair, Oberley, & Ho, 1991; Dong et al., 2006; Elroy-stein, Bernstein, & Groner, 1986). A rotenona é um insecticida inibidor do cI da mitocôndria. É utilizada em ratinhos com o intuito de produzir o fenótipo da DP, incluindo a degeneração dos neurónios dopaminérgicos, alterações motoras e a formação de inclusões fibrilares (Betarbet & Sherer, 2000). Ao contrario do MPTP, a rotenona é altamente lipofílica e, consequentemente, atravessa as membranas facilmente e entra nas células (Dauer & Przedborski, 2003). Deste modo, a rotenona poderia ter a capacidade de inibir o cI através do cérebro, ao atravessar a BHE. De facto, ratinhos sujeitos a uma administração crónica de rotenona desenvolveram uma degeneração nigral selectiva com a presença de α-Syn e inclusões similares aos CL, o que salienta a !. 28!.
(29) Introdução. susceptibilidade das células dopaminérgicas a alterações na mitocôndria (Betarbet & Sherer, 2000). Têm sido realizados diversos estudos, em diferentes áreas geográficas, na tentativa de estabelecer uma ligação entre o uso de pesticidas e o risco acrescido de DP. No entanto, a conclusão generalizada a que se chega é que apesar da DP poder estar associado ao uso de pesticidas a longo termo, não existe uma associação comprovada a nenhum componente em particular (Berry, La Vecchia, & Nicotera, 2010). Tabaco e Café O risco de vir a adquirir DP decresce cerca de 60% em fumadores e à volta de 30% em consumidores de café. Considera-se que este efeito neuroprotector se deva à nicotina e à cafeína respectivamente (Logroscino, 2005). O tabaco é um dos factores de risco mais estudados e um dos poucos em que se obteve resultados concretos. Diversos estudos demonstraram um risco inversamente proporcional entre o consumo de tabaco e a incidência de DP. Assim, têm sido propostos diversos mecanismos com o intuito de explicar o suposto efeito neuroprotector do tabaco. As explicações mais plausíveis sugerem o envolvimento da nicotina, devido às suas propriedades antioxidantes, a capacidade de estimular a libertação de dopamina e a acção inibidora sobre a iMAO-B, que activa a neurotoxicidade induzida pelo MPTP (Lau & Breteler, 2006). Em relação ao consumo de café existem evidências que a cafeína exerce um efeito protector no sistema dopaminérgicos. Um estudo realizado em 2001 demonstrou que ratinhos tratados com cafeína antes da exposição ao MPTP sofreram uma perda inferior de neurónios dopaminérgicos relativamente aos ratinhos sem tratamento prévio com cafeína. Pensa-se que o efeito neuroprotector da cafeína deve-se à acção inibitória desta sobre os receptores da adenosina-2 (Logroscino, 2005).. 29!.
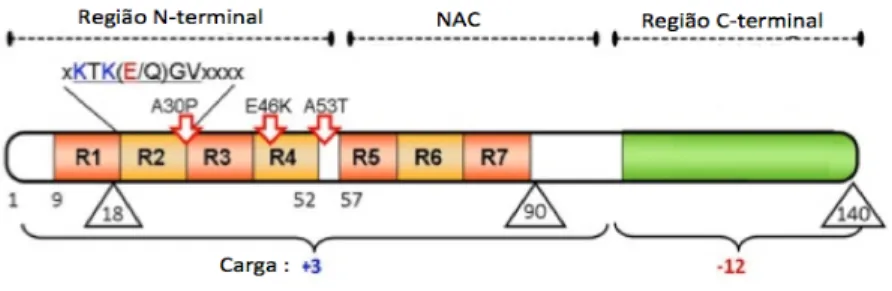
(30) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. ! 1.1.4.2. Factores Genéticos. A DP sempre foi considerada esporádica na natureza, contudo, nas últimas duas décadas, foram realizados inúmeros estudos genéticos, em diferentes áreas geográficas, que suportam a existência de uma substancial componente genética na DP. O primeiro gene a ser relacionado com a DP foi o SNCA, foi identificado numa família italiana que apresentava uma transmissão autossómica dominante. Deste então, foram identificados cerca de 18 loci relacionados com a DP (Bekris, Mata, & Zabetian, 2010).. Principais genes envolvidos na Doença de Parkinson PARK-1 e PARK-4 : Alfa-Sinucleína O locus PARK-1, juntamente com o locus PARK-4, contêm o gene SNCA, localizado no cromossoma 4q21-q23, que codifica para a α-Syn (Bekris et al., 2010). Até a data foram identificadas cinco mutações missenses : A53T, A30P, E46K, H50G e G51A, assim como duplicações e triplicações do gene wild-type (WT) (Trinh & Farrer, 2013). As mutações no gene SNCA são associadas à DP autossómica dominante de início relativamente precoce e rápida progressão, sendo um fenótipo mais agressivo e com elevada incidência de demência e danos cognitivos (Xiromerisiou & Dardiotis, 2010). A substituição A53T foi a primeira a ser identificada numa família com a forma autossómica dominante da DP, apresentando um declínio cognitivo e demência. Mais tarde foram identificadas as substituições A30P e E46K em duas famílias que apresentavam demência com CL (Bekris et al., 2010). Recentemente foram identificadas as mutações H50Q e G51A. A primeira foi identificada num caso esporádico de DP de início precoce e rápida progressão com declínio cognitivo e motor, enquanto a G51A foi identificada em 4 pacientes com DP de início precoce, rápida progressão com sintomas piramidais envolvidos (Appel-Cresswell et al., 2013; Lesage et al., 2013; Proukakis, Dudzik, & Brier, 2013). Posteriormente, descobriu-se que as duplicações e as triplicações do locus do gene SNCA podem causar PD autossómica dominante de início precoce. A idade de início das manifestações da DP parece estar relacionado com o número de copias dos genes !. 30!.
(31) Introdução. (Ikeuchi, Kakita, & Shiga, 2008). No caso dos pacientes que possuem uma duplicação neste locus, ostentando 3 copias deste gene, tendem a desenvolver DP por volta dos 50 anos, com um fenótipo mais típico, enquanto que os pacientes com triplicações, produzindo assim 4 copias do gene, desenvolvem a DP mais cedo, por volta dos 30 anos, com tendência a ser acompanhada por demência e CL difusos (Ikeuchi et al., 2008; Wood-Kaczmar, Gandhi, & Wood, 2006). Pode-se dizer, então, que a expressão aumentada de α-Syn parece ser tóxica para os neurónios, havendo uma relação doseefeito demonstrada pelas diferenças consoante o tipo de mutação (duplicação ou triplicação) (Wood-Kaczmar et al., 2006). PARK 2 : Parkina O Locus PARK 2 localiza-se no cromossoma 6q25.2-q27 e inclui o gene Parkina que codifica para a proteína com o mesmo nome (Bekris et al., 2010; Corti et al., 2011). Esta mutação foi originalmente identificada em famílias japonesas com parkinsonismo juvenil e uma transmissão autossómica recessiva (Farrer, 2006). A Parkina é uma enzima E3 que actua no processo de ubiquitinação como ligase ubiquitina-proteina, as mutações desta proteína levam alterações no SUP conduzindo a uma eliminação insuficiente do substracto e consequente agregação proteica (Corti et al., 2011) Clinicamente, este fenótipo manifesta-se muito precocemente, por volta dos 20 anos, mas também é frequente por volta dos 20-35 anos, sendo raro manifestar-se após os 50 anos (Corti et al., 2011). A progressão da doença é geralmente lenta e acompanhada de discinesia, distonia e alterações psiquiátricas (Bonifati, 2007). PARK- 5: UCL-L1 Localizado no cromossoma 4p14, este locus contém o gene UCH-L1 (Hidrolase L1 Ubiquitina Carboxiterminal) (Wirdefeldt, Adami, Cole, Trichopoulos, & Mandel, 2011). A UCL-L1 é uma enzima neuronal específica e uma das proteínas mais abundantes no cérebro, esta proteína está envolvida no sistema SUP e foi detectada em CL presentes nos neurónios dopaminérgicos da SNpc em doenças neurodegenerativas (Belin & Westerlund, 2008; Bonifati, 2007; Lowe, McDermott, Landon, Mayer, & Wilkinson, 1990).. 31!.
(32) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. ! Foi identificada uma mutação dominante da proteína UCH-L1 (193M) em dois membros de uma família afectada pela DP hereditária típica com início por volta dos 50 anos (Belin & Westerlund, 2008; Leroy, Boyer, & Polymeropoulos, 1998). Por outro lado, um polimorfismo na UCH-L1 (S18Y) aparenta ter um efeito protector, sugerindo uma redução no risco de desenvolver DP esporádica (Zhang et al., 2000). Liu et al. realizou um estudo em que propõe uma actividade de hidrólase e ligase, em simultâneo, para a proteína UCH-L1 (Liu, Fallon, Lashuel, Liu, & Lansbury, 2002). Deste modo, a sobre-expressão da mutação 193M da UCH-L1 provoca uma acumulação de α-Syn, sendo sugerido, também, que esta acumulação deve-se à ubiquitinação da α-Syn pela UCH-L1 dimerizada. Contudo, a variante polimórfica S18Y da UCH-L1 reduz a actividade ligase sem afectar a actividade de hidrólase da proteína, sendo uma possível explicação para o efeito protector deste polimorfismo (F. J. S. Lee & Liu, 2008; Liu et al., 2002). No entanto, permanece desconhecida a relação entre esta proteína e a neurodegeneração na DP familiar assim como o seu papel na DP esporádica (F. J. S. Lee & Liu, 2008). PARK-6 – PINK1 O Locus PARK-6 localiza-se no cromossoma 1p35-1p36 e possui o gene PINK1, que expressa para proteína com o mesmo nome (Corti et al., 2011). Pensa-se que a proteína-quinase PINK1 actua dentro da mitocôndria o que suporta a hipótese desta estar relacionada com a disfunção mitocondrial e com o stress oxidativo na patogénese da DP (Ross & Smith, 2007). A mutação no gene PINK1 foi identificada pela primeira vez numa família italiana e apresenta uma transmissão autossómica recessiva da DP, posteriormente foi também mapeada em oito famílias de diferentes países europeus com uma transmissão igualmente autossómica recessiva (Vila & Przedborski, 2004). Relativamente ao fenótipo clínico, apresenta um início precoce em que os primeiros sintomas manifestam-se por volta da quarta e quinta década de vida. As características clínicas assemelham-se à DP de início tardio, com uma progressão lenta, boa resposta à LDopa e, em alguns casos, demência e distúrbios psiquiátricos (Bekris, Mata, & Zabetian, 2010).. !. 32!.
(33) Introdução. PARK-7 : DJ-1 Este locus contém o gene DJ-1 e localiza-se no cromossoma 1p36 (Bekris et al., 2010). A proteína DJ-1 é expressa em diferentes tecidos humanos incluindo no cérebro, localiza-se no citoplasma, no núcleo e na mitocôndria das células (Ariga et al., 2013; Vila & Przedborski, 2004). É uma proteína multifuncional assumindo funções como chaperona, antioxidante, reguladora da transcrição, protease, regulação da mitocôndria e actua, também, como um sensor de stress sendo a sua expressão aumentada em situações de stress oxidativo. Devido às suas funções, acima descritas, esta proteína desempenha uma função de protecção contra a morte celular induzida pelo stress oxidativo (Ariga et al., 2013; Bekris et al., 2010; Farrer, 2006; Hardy, Lewis, Revesz, Lees, & Paisan-Ruiz, 2009; Vila & Przedborski, 2004). Pensa-se que a perda ou redução das funções desta proteína pode desencadear o início de doenças relacionadas com o stress oxidativo, nomeadamente a DP (Ariga et al., 2013). O gene DJ-1 foi primariamente identificado como um oncogene (Nagakubo et al., 1997) e posteriormente Bonifati et al. mapeou duas mutações em dois pacientes com DP, conduzindo à identificação do DJ-1 como um gene relacionado com a DP familiar de transmissão autossómica recessiva (Bonifati et al., 2003) As mutações no gene DJ-1 são responsáveis pela forma autossómica recessiva da doença e são pouco comuns, sendo responsáveis por 1-2% dos casos de DP com início precoce (Bonifati, 2007). Relativamente às características clínicas o conhecimento é limitado, sabe-se que apresenta um início precoce (30-40 anos) e uma progressão lenta, alguns pacientes manifestam quadros psiquiátricos (Bekris et al., 2010; Corti et al., 2011). PARK-8 : LRRK2 O locus PARK8 encontra-se no cromossoma 12p21 e contém o gene que expressa a proteína LRRK2 tendo sido identificado pela primeira vez numa família japonesa e apresenta uma transmissão autossómica dominante (Belin & Westerlund, 2008). A LRRK2 é uma proteína com funções de quinase e GTPase e actua, também, em múltiplos processos biológicos como: transmissão neuronal, arborização neuronal, endocitose, autofagia e imunidade(Trinh & Farrer, 2013). Até aos dias de hoje, as mutações dominantes missense na LRRK2 são a causa genética mais comum de DP e, de acordo com os estudos disponíveis, são responsáveis por cerca de 10 % dos casos de DP familiar autossómica dominante. 33!.
(34) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. ! (Bonifati, 2007; Kett et al., 2012). Contudo, apesar de já terem sido reportadas diversas variantes, apenas seis delas são consideradas patogénicas (R1441G, R1441C, R1441H, Y1699C, G2019S e I2020T) (Bekris et al., 2010). Um aspecto interessante relativamente a este gene, é o facto da mutação Gly2019Ser apresentar uma frequência elevada não só em casos de DP familiar como também em casos de DP esporádicos, em diversas populações, sendo a mutação mais comum (Bekris et al., 2010; Bonifati, 2007; Hardy et al., 2009). O fenótipo clínico apresenta elevada analogia com a DP idiopática, o início dos sintomas ocorre por volta dos 60 anos, contudo, já foram reportados casos de início precoce e de início tardio, abrangendo idades entre os 20 e os 90 anos (Gasser, 2009; Ross & Smith, 2007). Os portadores das mutações no gene LRRK2 aparentam ter uma DP mais benigna visto que é de progressão lenta e apresenta uma boa resposta ao tratamento com L-Dopa e uma frequência diminuta de demência e outras complicações psiquiátricas (Bekris et al., 2010; Gasser, 2009; Ross & Smith, 2007).. ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! !. !. 34!.
(35) Introdução. Tabela 1 – Resumo dos Genes Envolvidos na DP ( Adaptado de Bekris et al., 2010; Corti et al., 2011; Farrer, 2006; Gasser, 2009; Trinh & Farrer, 2013; Vila & Przedborski, 2004; Wirdefeldt et al., 2011). Locus. Cromossoma. Gene. Herança. PARK-1 PARK-4. 4q21-q23. SNCA. Dominante. PARK-2. 6q25.2-q27. Parkina. Recessiva. PARK-3. 2p13. --. Dominante. PARK-5. 4p14. UCHL-1. Dominante. PARK-6. 1p35-p36. PINK1. Recessiva. PARK-7. 1p36. DJ-1. PARK-8. 12p12. LRRK2. Dominante. PARK-9. 1p36. ATP13A2. Recessiva. PARK-10 PARK-11 PARK-12 PARK-13. 1p32 2q37.1 Xq21-q25 2p13. -GIGYF2 -OMI/HtrA2. PARK-14. 22q13.1. PLA2G6. PARK-15. 22q12-q13. FBX07. PARK-16. 1q32. --. Recessiva. 35!. Fenotipo Início Relativamente precoce Progressão rápida Boa resposta a L-dopa Disfunção cognitiva precoce Início Precoce Juvenil Progressão lenta Distonia e Discinesia Início 50 anos Boa resposta a L-dopa Disfunção cognitiva Início 50 anos Tremor inicial antecede a bradicinesia Boa resposta a L-dopa Início precoce Progressão Lenta Boa resposta a L-dopa Início precoce de discinesias induzidas por fármacos Início precoce 30 anos Progressão Lenta Boa resposta a L-dopa Pode ocorrer alterações psiquiátricas Início 40-50 anos Boa resposta a L-dopa Demência ocasional Início Precoce Progressão rápida Sintomas Piramidais Demência. Dominante Dominante -Dominante Início Tardio Início Precoce Recessiva Distonia Sintomas piramidais Recessiva Início Precoce Sintomas Piramidais --.
(36) Mecanismos Moleculares envolvendo a alfa-Sinucleína e a Sinflina-1 na Doença de Parkinson. !. 2. Diagnóstico e Farmacoterapia na Doença de Parkinson 2.1 Quadro Clínico e Diagnóstico da DP Clinicamente, a DP é definida pela presença de sinais motores cardinais: tremor, rigidez, bradicinesia e instabilidade postural. Contudo, a ocorrência de sintomas não motores como insuficiência autónoma, danos cognitivos, depressão, deficiências olfactivas, psicose e distúrbios do sono são bastante comuns durante o decurso da DP (Jankovic, 2006; Samii, Jg, & Br, 2004). O início dos sintomas normalmente é unilateral e dissimulado, havendo uma progressão lenta mas conservando-se o carácter unilateral. Os primeiros sinais são vagos, pode haver uma sensação de dor, dormência, fraqueza, dores musculares e rigidez. Pode ocorrer, como sinal inicial, um tremor num membro, geralmente quando o doente está em repouso que aumenta com as emoções e desaparece durante o sono (Guimarães & Alegria, 2004). O diagnóstico clínico é tipicamente baseado na presença dos sintomas motores cardinais, a ausência de sintomas atípicos sugestivos de um diagnóstico alternativo e a resposta ao tratamento com L-Dopa. Os critérios clínicos de diagnóstico mais usados baseiam-se nos apresentados pela UK PD Society Brain Bank (Tabela 2). Uma das complicações no diagnóstico da DP é a distinção entre outras síndromes parkinsonianas, como as Atrofias Multissistémicas e a Paralesia Supranuclear Progressiva, nos estágios iniciais da doença (Brooks, 2010; Samii et al., 2004).. !. 36!.
(37) Diagnóstico e Farmacoterapia na Doença de Parkinson. Tabela 2 - Critérios de Diagnóstico da DP (Adaptado de UK PD Society Brain Bank Clinical Diagnostic Criteria (Hughes, Daniel, Kilford, & Lees, 1992) ). Fase 1: Diagnóstico de sindroma parkinsónica: ¬ Bradicinesia ¬ Mais pelo menos 1 dos seguintes: • Rigidez muscular • Tremor de repouso a 4-6Hz • Instabilidade postural Fase 2: Critérios de exclusão de DPI: • • • • • • • • • • • • • • •. História de AVC’s repetidos com progressão em escada das manifestações parkinsónicas História de traumatismos cranianos repetidos História de encefalite Crises oculogíricas Tratamento neuroléptico no início das manifestações Remissão sustentada Persistência de manifestações unilaterais após 3 anos de evolução Paralisia supranuclear do olhar Sinais cerebelosos Desenvolvimento precoce de sintomas autonómicos Desenvolvimento precoce de demência Sinal de Babinski Presença de tumor cerebral ou de HPN Resposta negativa a doses elevadas de L-Dopa (excluída a má absorção) Exposição ao MPTP. Fase 3: Critérios adicionais que apoiam o diagnóstico de DPI: • • • • • • • •. Início unilateral Tremor de repouso presente Carácter progressivo Persistência da assimetria afectando mais o lado de início Resposta excelente (70 a 100%) à L-Dopa Movimentos coreicos induzidos pela L-Dopa Resposta à L-Dopa durante pelo menos 5 anos Evolução clínica durante mais de 10 anos. 37!.
Imagem


+7





Documentos relacionados
Para o Planeta Orgânico (2010), o crescimento da agricultura orgânica no Brasil e na América Latina dependerá, entre outros fatores, de uma legislação eficiente
b) Execução dos serviços em período a ser combinado com equipe técnica. c) Orientação para alocação do equipamento no local de instalação. d) Serviço de ligação das
O objetivo deste estudo foi determinar a prevalência da coccidiose em frangos de corte no ano de 2012 em uma integração localizada na região noroeste do estado do
Os interessados em adquirir quaisquer dos animais inscritos nos páreos de claiming deverão comparecer à sala da Diretoria Geral de Turfe, localizada no 4º andar da Arquibancada
E mais: é possível a coexistência de mais de um estilo de época em um mesmo período de tempo, principal- mente quando há uma transição de valores, ou seja, há uma mistura de
Es importante poner de relieve que la magna carta brasileña, en materia de propiedad y orden económica, artículo 170, garantiza el derecho de propiedad a empresas en su apartado
Figura 9- Padrão de terminação dos refletores das unidades 1 a 4 em onlap com o Alto Cabo Frio e reflexões descontínuas no limite inferior da unidade oligocênica. Na linha
segunda guerra, que ficou marcada pela exigência de um posicionamento político e social diante de dois contextos: a permanência de regimes totalitários, no mundo, e o
