REVIEW
Stem and progenitor cell-mediated tumor selective
gene therapy
KS Aboody
1,2, J Najbauer
1and MK Danks
31Division of Hematology/Hematopoietic Cell Transplantation, City of Hope National Medical Center and Beckman Research Institute,
Duarte, CA, USA;2Division of Neurosciences, City of Hope National Medical Center and Beckman Research Institute, Duarte, CA, USA
and3Department of Molecular Pharmacology, St Jude Children’s Research Hospital, Memphis, TN, USA
The poor prognosis for patients with aggressive or metastatic tumors and the toxic side effects of currently available treatments necessitate the development of more effective tumor-selective therapies. Stem/progenitor cells display inherent tumor-tropic properties that can be exploited for targeted delivery of anticancer genes to invasive and metastatic tumors. Therapeutic genes that have been inserted into stem cells and delivered to tumors with high selectivity include prodrug-activating enzymes (cytosine deaminase, carboxylesterase, thymidine kinase), interleukins (IL-2, IL-4, IL-12, IL-23), interferon-b, apoptosis-promoting genes (tumor necrosis factor-related apoptosis-inducing ligand) and metalloproteinases (PEX). We and others have demonstrated that neural and mesenchymal stem cells can deliver therapeutic genes to elicit a significant antitumor
response in animal models of intracranial glioma, medullo-blastoma, melanoma brain metastasis, disseminated neuro-blastoma and breast cancer lung metastasis. Most studies reported reduction in tumor volume (up to 90%) and increased survival of tumor-bearing animals. Complete cures
have also been achieved (90% disease-free survival for41
year of mice bearing disseminated neuroblastoma tumors). As we learn more about the biology of stem cells and the molecular mechanisms that mediate their tumor-tropism and we identify efficacious gene products for specific tumor types, the clinical utility of cell-based delivery strategies becomes increasingly evident.
Gene Therapy (2008) 15, 739–752; doi:10.1038/gt.2008.41; published online 27 March 2008
Keywords:neural stem cells; mesenchymal stem cells; targeted tumor therapy; enzyme/prodrug therapy; malignant tumors; cancer treatment
Introduction
Stem and progenitor cell-mediated gene delivery is emerging as a strategy to improve the efficacy and minimize the toxicity of current gene therapy ap-proaches. Multiple potential sources for clinically useful stem and progenitor cells have been identified, including autologous and allogeneic embryonic, fetal and adult somatic cells from neural, adipose and mesenchymal tissues. In the case of regenerative medicine, trans-planted stem and progenitor cells themselves comprise the therapeutic modality by replacing or regenerating damaged tissue. In the case of enzyme or other genetic deficiencies, these cells may engraft and express the deficient enzyme or gene product. A third potential use for stem and/or progenitor cells, and the topic of this review, is their use as delivery vehicles for therapeutic gene products. Current evidence indicates that systemi-cally administered stem/progenitor cells migrate to and
infiltrate primary and metastatic solid tumors, and that these cells can be used to deliver therapeutic genes, cDNAs or siRNAs selectively to tumor foci. We and others envision that this inherent tumor-tropism of stem/progenitor cells can be exploited to develop effective, well-tolerated treatments for patients with malignant solid tumors.
Neural and mesenchymal stem/progenitor cells, iso-lated from various tissue sources, possess a remarkable inherent tumor tropism. The major advantage in using stem/progenitor cells as delivery vehicles lies in their potential to achieve a heretofore unachievable therapeu-tic specificity. Neural stem/progenitor cells, and mesenchymal stem cells (MSCs) harvested from bone marrow or adipose tissue, for example, display tumor-tropic properties in preclinical models of malignant primary and metastatic tumors. This tropism has been exploited to deliver multiple therapeutic genes or cDNAs selectively to tumor foci (summarized in Table 1; stem and progenitor cells for tumor-selective therapy).3,10,15–17,23–31
The central question to be addressed then becomes whether we can exploit the inherent ability of these cells to migrate to, infiltrate and—when desirable—engraft to ‘correct’ pathologies in patients. The long-range goal is complete cure without toxic side effects or relapse. The principal criteria that will determine the clinical utility of these strategies are safety, feasibility and efficacy. This review will focus on the advantages of neural stem/
Received 14 February 2008; accepted 16 February 2008; published online 27 March 2008
Correspondence: Dr KS Aboody, Divisions of Hematology/Hema-topoietic Cell Transplantation, City of Hope National Medical Center and Beckman Research Institute, 1500 E Duarte Road, Duarte CA 91010, USA.
E-mail: kaboody@coh.org and Dr MK Danks, Department of Molecular Pharmacology, St Jude Children’s Research Hospital, 332 N Lauderdale, Memphis, TN 38105, USA.
E-mail: mary.danks@stjude.org
Gene Therapy (2008) 15, 739–752
&2008 Nature Publishing Group All rights reserved 0969-7128/08$30.00
Table 1 Genetically modified stem cells demonstrate tumor targeting and/or therapeutic efficacy in preclinical models of primary and secondary brain tumors, melanoma brain metastasis, breast carcinoma metastasis to lung, and disseminated neuroblastoma
Neural stem cells type (source)
Route of administration
GLIOMA (source) and location
Tumor host Vector Therapeutic
transgene
Therapeutic efficacy
References
C17.2 (mouse) Intratumoral CNS-1 (rat glioma) Intracranial
Nu/numouse HSV-1 vector HSV-1 vector Not tested Herrlingeret al.1
C57.npr.IL-4 (mouse)
Intratumoral GL261 (mouse glioma) Intracranial
C57BL/6J mouse Retrovirus IL-4 Extended survival Benedettiet al.2
ST14A.IL-4.3 (rat) Intratumoral C6 (rat glioma) Intracranial
Sprague–Dawley rat Retrovirus IL-4 Extended survival Benedettiet al.2 C17.CD2
(mouse)
Intracranial -C17.2 mixed with CNS-1
CNS-1 (rat glioma) Intracranial
Nu/numouse Retrovirus CD (5-FC i.p.) B80% reduction in
tumor volume
Aboodyet al.3
NSC-IL-12 (mouse) Intratumoral GL261 (mouse glioma) Intracranial
Sprague–Dawley rat Adenovirus IL-12 Extended survival Ehteshamet al.4
NSC-TRAIL (mouse)
Intracranial/ intratumoral
U343MG (human glioma)
Intracranial
Nu/numouse Adenovirus TRAIL Tumor size
reduction
Ehteshamet al.5
ST14A (rat) Intracranial -ST14A mixed with C6 cells
C6 (rat glioma) Intracranial
Sprague–Dawley rat Retrovirus CD (5-FC i.p.) B50% reduction in tumor growth
Barresiet al.6
C17.2, C17.CD2 (mouse)
Intravascular CNS-1 (rat glioma) U-251 (human glioma) Intracranial,
Subcutaneous
Nu/numouse Retrovirus N/A Distribution study
NPC-FLsTRAIL (mouse)
Intracranial Gli36-RL (human glioma)
Intracranial
Nu/numouse pKSR2–Herpes
simplex virus type 1 amplicon
S-TRAIL 480% reduction in tumor growth
Shahet al.7
HB1.F3-PEX (human)
Intracranial U87MG (human glioma) Intracranial
Swiss nude mouse PTracer-BsdPEX vector
PEX 90% reduction in
tumor volume
Kimet al.8
BM-NSC-IL23 (mouse)
Intracranial GL261 (mouse glioma)
C57BL/6,Nu/nu, CD4 T-cell KO
IL-23 IL-23 Extended survival Yuanet al.9
C17.CD2 (mouse) Intracranial B16/F10 (mouse melanoma) Intracranial
C57BL/6J mouse Retrovirus CD (5-FC i.p.) B70% reduction in tumor volume
Aboodyet al.10
HB1.F3 (human) Intracranial U251, D566 (human glioma) Intracranial
Nu/numouse N/A N/A Distribution study Linet al.11
NSCtk (rat) Intracranial (ipsi- or contralateral to tumor)
C6 (rat glioma) Intracranial
Sprague–Dawley rat HSV Thymidine kinase (Ganciclovir, i.p.)
B90 and 70% reduction in tumor volume, ipsi- or contalateral injection, respectively; extended survival
Liet al.12
Skin-derived stem cells—hSDSC (human)
Intracranial (ipsi- or contralateral to tumor)
U87MG (human glioma) Intracranial
Nu/numouse N/A N/A Reduction in tumor
volume, extended survival
Liet al.12
Cell-mediated
anticancer
therapy
KS
Aboody
et
al
Neural stem cells type (source)
Route of administration
Medulloblastoma (source) location
Tumor host Vector Therapeutic
transgene
Therapeutic efficacy
References
HB1.F3.CD (human) Intratumoral Daoy (human medulloblastoma) Intracranial
Nu/numouse Retrovirus CD (5-FC i.p.) 76% reduction
in tumor volume
Kimet al.13
HB1.F3.CD (human) Intracranial (cisterna magna)
Daoy (human medulloblastoma) Intracranial
Nu/numouse Retrovirus CD (5-FC i.p.) Extended
survival
Shimatoet al.14
Neural stem cells type (source)
Route of administration
Melanoma (source) location
Tumor host Vector Therapeutic
transgene
Therapeutic efficacy
References
C17.CD2 (mouse) Intravascular B16/F10 (mouse melanoma) Intracranial (intracarotid inj)
C57BL/6J mouse Retrovirus CD (5-FC i.p.) B70% reduction in tumor volume
Aboodyet al.10
HB1.F3.CD (human)
Intracranial (cisterna magna)
Daoy (human medulloblastoma) Intracranial
Nu/numouse Retrovirus CD (5-FC i.p.) Extended
survival
Shimatoet al.14
Neural stem cells type (source)
Route of administration
Neuroblastoma (source) location
Tumor host Vector Therapeutic
transgene
Therapeutic efficacy
References
HB1.F3.rCE (human)
Intravascular (tail vein)
NB-1691, NB-1643, SK-N-AS (human neuroblastoma) Disseminated
Es1e SCID mice Adenovirus rCE (CPT-11 i.v.)
100% tumor-free survival for
46 months
Aboodyet al.15
HB1.F3.rCE (human)
Intravascular (tail vein)
NB-1691, NB-1643, SK-N-AS
(human neuroblastoma) Disseminated
Es1e SCID mice Adenovirus rCE (CPT-11 i.v.)
90% tumor-free survival for
41 year
Dankset al.16
HB1.F3. IFN-b
(human)
Intravascular (tail vein)
NB-1691 (human neuroblastoma) Disseminated
Es1e SCID mice Adenovirus IFN-b Significant tumor reduction/slowed disease progression
Dicksonet al.17
Mesenchymal stem cells type (source)
Route of administration
Glioma (source) location
Tumor host Vector Therapeutic
transgene
Therapeutic efficacy
References
MSCs (rat) Intracranial/ Intratumoral or Contralateral
9L (rat glioma) Fisher rat Adenovirus IL-2 90% reduction in
tumor volume; Extended survival
Nakamuraet al.18 MSCs (human) Intratumoral/
Intracarotid
U87 (human glioma)
Nu/numouse Adenovirus IFN-b Extended survival Nakamizoet al.19
MSCs (BM-TIC-tk-GFP) (rat)
Intratumoral 9L (rat glioma)
Fisher rat HSV-tk39 Thymidine kinase (Ganciclovir, i.p.)
Extended survival Mileticet al.20
Table 1 Continued
Cell-mediated
anticancer
therapy
KS
Aboody
et
al
741
Gene
progenitor cells as delivery vehicles for tumor-selective therapy in models of primary and metastatic cancer. We will discuss the sources of stem and progenitor cells available, the types of ‘therapeutics’ that might be delivered, approaches to engineering the cells to express these genes and factors that will increase the likelihood that cell-based delivery systems will be clinically useful.
Tumor tropism and infiltrative potential
Based on their unique tropism to sites of pathology, the principal advantage of stem/progenitor cell-based delivery of anticancer therapeutics is in the potential to increase the tumor-specificity and therefore the therapeutic index of therapies of interest. Importantly, and in striking contrast to virtually all other delivery approaches, neural stem/progenitor cells migrate to and infiltrate bulk tumorsin vivo, and target invasive tumor cells (Figure 1). This ability to localize to and distribute through tumor masses would be predicted to mediate a more complete and robust antitumor response than vehicles with more restricted distribution potential such as modified liposomes,32–34conjugated antibodies, tumoror organ-specific expression cassettes, nanoparticles or polymers,35–37or modified viruses of various genotypes
(reviewed in Waehler et al.38). While each approach
undoubtedly has advantages and disadvantages, cell-based approaches compare favorably with all approaches currently being investigated. Targeted liposomes, antibodies or modified viruses rely on interaction of proteins or peptides with ‘receptors’ on the surface of targeted tumors or organs. The presump-tion that must be made with these types of targeted approaches is that proteins (or other surface molecules) can be identified, which are sufficiently unique in level or pattern of expression to serve as ‘specific receptors’ for the targeting moiety. In antitumor approaches, for example, numerous constructs have been reported that target transferrin receptor,39–45 since this receptor has
been reported to be expressed at levels approximately twofold higher in tumors than in normal tissues.46,47
Intuitively, a twofold difference may not provide a sufficient differential between tumor and normal tissue to confer tumor selectivity, unless the dose-response curve of the therapeutic entity is very steep.
The tumor-tropism of cell-based delivery is likely mediated by multiple cell-surface and secreted proteins, and does not depend exclusively on the expression of a single protein or ‘receptor’. While the molecular basis for the tumor-tropism of stem/progenitor cells in vivo is poorly understood, it has been observed that numerous cytokines, growth factors and receptors modulate migration in vitro. Candidate cytokine/receptor pairs include SDF-1/CXCR4,48 SCF/c-Kit,49,50 HGF/c-Met,51
VEGF/VEGFR,52MCP-1/CCR253and HMGB1/RAGE.54,55
Among adhesion molecules, b1- and b2-integrins, and L-selectin may play a significant role in the mobilization and homing of stem cells.56–58 Extracellular matrix
proteins, and MMP2, MT1-MMP and TIMP-2 have also been associated with the tropism of stem/progenitor cells.59,60However, the molecular mechanisms of stem/
progenitor migration to tumors of various origins and phenotypes have yet to be elucidated.
Stem and progenitor cell survival
in vivo
A second advantage to cell-based delivery is that cells can be manipulated to survive long- or short termin vivo. Clearly, individual applications will determine which alternative is more appropriate. Correction of a genetic enzyme deficiency using bone marrow MSCs or neural stem/progenitor cells engineered to express a functional human enzyme would require long-term delivery cell survival—either resulting from engraftment or from fusion with endogenous cells. For chemotherapy appli-cations, delivery cells need to survive only sufficiently long to mediate effective therapy. This type of approach is exemplified by cell-based delivery of prodrug-activating enzymes such asHerpes simplexvirus thymidine kinase, to activate (phosphorylate) ganciclovir, Escherichia colicytosine deaminase (CD) to convert 5-fluorocytosine (5-FC) to 5-fluorouracil, or carboxylesterase (CE) to convert CPT-11 (irinotecan) to SN-38.20,61–68We have found that in
the absence of anticancer drug treatment, neural stem/progenitor cells remain viable, at least 2–3 weeks following intratumoral injection into mice bearing
orthotopic gliomas. Similarly, other laboratories have noted stem cell survival of 2–12 weeks in preclinical models of cerebral ischemia/stroke, Parkinson’s disease and Huntington’s disease.69–73Interestingly, in the latter
type of pathological environments, neural stem/pro-genitor cells have been reported to differentiate into neurons and/or glial cells over time. In contrast, in mice that have received drug treatment as a component of stem cell/enzyme/prodrug tumor therapy for neuro-blastoma, we found no evidence of long-term stem cell survival16 and unpublished observations). Of note,
neural stem/progenitor cells injected into the brain of normal immunocompetent or immunocompromised mice begin to undergo apoptosis within 72 h (KS Aboody
et al., unpublished data). There is some evidence that a
subpopulation of stem or progenitor cells fuse with endogenous cells,74–76but our studies with immortalized
neural stem/progenitor cell lines have not corroborated this finding. Fusion may be characteristic of the particular stem/progenitor cells or cell line used. It has also been reported that MSCs engraft into the stroma of tumors, resulting in persistence of the clonal population of exogenous stem cells.19,26,77,78The critical concern for
all approaches will be the careful design of vectors and characterization of the specific stem/progenitor cells, to ensure that their duration of survival is appropriate to the specific clinical application of interest. A Phase 1 neural stem cell (NSC)-mediated glioma therapy clinical trial is currently under development (Aboody and Portnow, City of Hope National Medical Center). This trial includes biologic studies to determine the character-istics of NSC persistence, fate and distribution in the human brain.
Tissues targeted by cell-based delivery
vehicles and cellular localization of
delivered transgenes
A third advantage of stem/progenitor cells compared to other types of delivery vehicles, is the ability of these cells to migrate to tumor foci regardless of tumor size, anatomic location or tissue of origin.30 When
adminis-tered intracranially or intravenously, cloned NSC lines, for example, migrate to orthotopic gliomas,3,8
medullo-blastoma10,14 and melanoma brain metastases10 in
pre-clinical models. The unique characteristic of efficiently crossing the blood-brain barrier provides a very attrac-tive advantage in treating central nervous system (CNS)-associated tumors. Intravenously administered NSCs also migrate to disseminated neuroblastoma tumors in multiple anatomic locations, including the liver, ovaries and bone marrow.15,16 NSCs also migrate to
subcuta-neous xenografts in mice bearing tumors from human breast, prostate and melanoma tumor cell lines.10,21,26,30,79
These observations suggest that the potential for clinical application of cell-mediated delivery of therapeutic transgenes may be quite extensive. For each therapeutic application, however, it will also be important to be cognizant of molecular processing required, once cell-mediated delivery of a therapeutic has reached the target site. An antibody-drug conjugate, for example, would be endocytosed into cells and sequestered within endo-somes in the cytoplasm. A curative amount of active Figure 1 Neural stem cells (NSCs) infiltrate tumor foci and also
target invading tumor cells. (a) NSCs (x-gal labeled blue), when administered directly into tumors in a preclinical orthotopic glioma model, distribute through bulk tumor mass (black arrowheads) and colocalize with invasive tumor cells (red). (b) NSCs (blue) can ‘track’ single invasive tumor cells (red) infiltrating into normal tissue away from primary tumor mass. Based on Figure 2 from Aboody et al.,3 with permission from the National Academy of Sciences (USA).
Cell-mediated anticancer therapy KS Aboodyet al
743
drug must then traffic from the endosome to the intracellular organelle that is sensitive to the cytotoxic effect of the drug. In comparison, cells as delivery vehicles may not be subject to endosomal compartmenta-lization, but other characteristics must be considered, such as how efficiently the prodrugs or activated forms of these drugs cross cell membranes and how the permeability characteristics of each form of the drug might affect overall antitumor efficacy. Details of specific components of individual therapeutic approaches not-withstanding, the general abilities of stem and progeni-tor cells to traverse the blood brain barrier and to migrate to multiple types and sizes of tumors in various anatomical locations are obvious advantages of cell-based delivery vehicles.
Availability of stem and progenitor cells
As we become more proficient at harvesting and expanding appropriate stem and progenitor cellsin vitroor at de-differentiating abundant somatic cells,80–83 cell
number will become less of an issue. Methods are also being developed to harvest stem cells from organs other than those commonly used thus far. In addition to bone marrow, umbilical cord, adipose tissue and brain, recent reports indicate that dental pulp,84 first trimester fetal
blood85 or embryonic endothelial cells86 may also be
sources of therapeutically useful stem or progenitor cells. Currently, however, while standard methods are avail-able to produce virtually unlimited quantities of anti-bodies, viruses, liposomes or PEGylated nanoparticles, culture methods have not yet been perfected to rapidly produce unlimited quantities of undifferentiated stem and progenitor cells. The lack of adequate numbers of autologous cells, or the limited amount of time available to expand, modify and test them is currently a limitation of patient-derived cell-based delivery vehicles. Further-more, these pools of primary expanded cell cultures may change over time during passage, thereby making them extremely difficult to characterize or to establish master cell banks. We believe that this constraint is efficiently overcome by use of immortalized, clonal stem/progeni-tor cell lines. Following selection and clonal expansion, cell lines can be tested for stability, biodistribution, safety and toxicity. Well-characterized cell lines may then be expanded into master cell banks for clinical use.
Immortalized, clonal stem/progenitor
cell lines
Well-characterized cell lines of allogeneic origin have been used in preclinical models of glioma, medulloblas-toma, melanoma, neuroblastoma and breast carcinoma (Table 1) with some noteworthy successes. The use of well-characterized, immortalized allogeneic cell lines for clinical trials would circumvent problems of cell avail-ability and minimize potential insertion site-mediated toxicities (discussed below), but will make it necessary to evaluate the tumorigenic and immunogenic potentials of the cells themselves and of the vector used to engineer transgene expression. Readily available autologous stem cell sources would be ideal. Potential tissue sources for these cells have been suggested, with cells from each
source having unique properties. For example, Kern et al.87observed that MSCs from bone marrow had a lower
proliferation capacityin vitrothan MSCs from umbilical cord blood or adipose tissue, making stem cells from the latter two sources attractive alternatives to the thus far more frequently used bone marrow MSCs based on the consideration of cell number. However, the likelihood that adequate numbers of autologous cells will be available or can be generated by culturing for a limited
time in vitro is unknown and may depend on the cell
type being used. Further, even if adequate numbers of pathology-free autologous cells could be generated, the time required to genetically modify, expand, characterize and certify these cells might prohibit their use in patients with a limited life expectancy. The high cost and limited number of centers capable of producing such cells might well also be prohibitive. We propose that well-character-ized, cloned cell lines comprise clinically useful delivery vehicles.
Cell lines as delivery vehicles: toxicity
or the lack thereof—tumorigenicity and
immunogenicity
Toxicity/tumorigenicity
Preclinical data and results of early, cell-based gene therapy patient trials suggest that the most likely toxicity of cell-based gene therapies is the generation of secondary malignancies.88 Allogeneic cell lines used as
delivery vehicles could be predicted to be tumorigenic by three mechanisms: (1) expression of the gene used to immortalize the delivery cell line transforms the cells; (2) insertion of the immortalizing or therapeutic transgene at a critical locus in the genome dysregulates expression of other proteins and induce tumors and (3) the delivery cell fuses with endogenous cells, and the resulting fused cell has a transformed phenotype.
With respect to the first mechanism, several genes have been evaluated for their potential utility in immortalizing stem cells for use as delivery vehicles: the proto-oncogene v-myc,3,70,72,89–91the catalytic
compo-nent of the telomerase complex (human telomerase reverse transcriptase (hTERT)), human papillomavirus type 16 E6/E7 genes, Bmi-192–99 and an N-terminal
fragment of SV40 large T-antigen.2,100 Characteristics of
the resultant cell lines, each of which had extended life spans in culture, have been reviewed.101Pertinent to the
focus of the current review, several cell lines immorta-lized with hTERT and SV40 large T fragment were observed to be unstable with respect to chromosome number and karyotype. Additional evidence suggests that ectopic expression of telomerase in MSCs increases their resistance to radiation,102 whereas in NT2 neural
progenitors, hTERT inhibits neuronal differentiation.103
In contrast, the cell lines with which our laboratories have the most experience were immortalized using the
v-mycgene.70,72The multiple antitumor studies performed
with murine (C17.2) or human (HB1.F3) clonal popula-tions of v-myc-immortalized fetal neural cells are indicated in Table 1 and have been reviewed pre-viously.30 These cells do not form colonies in soft agar
(MK Danks, unpublished observations) and have not been observed to form tumors in SCID mice up to 12
months following intravenous or intracranial injection. Thus far, it appears that of the genes evaluated for immortalizing stem cell lines, v-myc appears most promising for generating cell lines with clinical utility.
With respect to the second potential mechanism of tumorigenesis, the critical nature of the genomic inser-tion site of genes used to immortalize cell lines or as components of therapy is well documented by a recent clinical trial. In this trial, patients with immune defi-ciencies received uncloned autologous hematopoietic stem cells that had been transduced with retrovirus. In a subset of patients, leukemogenic clonal populations of cells survived. As reviewed recently by Pike-Overzet
et al.,88the engrafted populations of hematopoietic cells
that survived had incorporated the exogenous nucleotide sequence at sites adjacent to loci encodingLMO2,LYL1,
c-Jun,Bmi1or CCND2. Insertions at these sites
upregu-lated expression of genes, which, when highly expressed, transformed T cells. The critical concern becomes, in any given cell type, if an exogenous gene or cDNA inserts into the host cell genome, is the expression of any ‘nearby’ gene altered and is upregulation of that gene likely to impact on the phenotype of the type of cell into which the gene was inserted? (It should be noted that cell lines immortalized using retroviruses to insert hTERT, SV40 large T, Bmi-1 and E6/E7 have not been extensively characterized. Since insertion sites were not mapped, hTERT itself may not be tumorigenic, but its insertion may have disrupted another critical genomic locus and transformed the delivery cell. Before telomerase-immor-talized cell lines are used clinically, it will be essential to determine which of these alternatives is responsible for any observed tumor development.) In leukocytes, un-fortunately, there is documentation from clinical trials that insertion of an exogenous gene near at least five specific genetic loci increases the tumorigenic potential. Based on these data, one would postulate that if cells of neuronal, rather than hematopoietic, origin were used as delivery vehicles it would be important to determine that insertion of exogenous sequences near genetic loci encoding MYC-N or EGFR, for example, did not alter expression of these genes. Pike-Overzet et al.88 suggest
that detrimental insertion sites likely differ in different cell types. The difficulty in characterizing all insertion sites in uncloned populations of primary transduced cells again emphasizes a potential advantage of cloned characterized cell lines compared to primary cells as vehicles, if therapeutic transgenes have been incorpo-rated into the delivery cell genome.
With respect to the third mechanism noted above, delivery cells could potentially become tumorigenic through fusion of the delivery cell with a tumor cell. This is least likely of the three potential mechanisms for multiple reasons. First, hyperdiploid cells have not been observed in animals receiving stem/progenitor cells. Second, Foroni et al.104 showed that expression of
exogenous c-myc immortalizes primary stem cells, but does not transform them. Importantly, these investiga-tors also showed that co-expression of mycand ras did not transform adult NSCs. Third, embryonic stem cells appear to have a greater capacity for fusion than fetal or adult cells,105,106and the HB1.F3 cells are of fetal origin.
Further, multiple studies found no evidence of bone marrow MSCs or NSC fusion with hepatocytes,
pancrea-tic endocrine cells, endothelial cells or epidermis.107–110
While high levels of specific cytokines can induce fusion of MSCs with cardiomyocytes or striated muscle cells,111,112it appears that nonembryonic stem cells have
little capacity to fuse with cells of a recipient organism. Possibly, fetal or adult stem or progenitor cells will have more utility than embryonic stem cells as delivery vehicles.
Immunogenicity
Primary neural stem and progenitor cells have been characterized by multiple laboratories as having low, if any, immunogenic potential in their undifferentiated state. Horiet al.113reported that CNS progenitor cells do
not express MHC class I or II antigens either at the time of harvest, upon culture in 10% serum or when grown as subcapsular renal grafts in vivo. Interestingly, in vitro
culturing of primary adipose-derived cells for 3–4 passages diminished their ability to stimulate prolifera-tion of allogeneic responder T-cells to below detectable levels. While a minimal-to-no immunogenic potential is certainly possible since, for example, the HB1.F3 cell line used in our laboratories expresses undetectable levels of MHC Class I antigens and extremely low levels of Class II antigens, the more likely probability is that reported by Aboody et al.3 These investigators showed that stem/
progenitor cells of neural origin elicit a subacute, limited, local immune response characterized by detectable T-cell infiltration, but with persistence of viable HB1.F3 human NSCs in syngeneic models of immunocompetent mice bearing syngeneic orthotopic gliomas. Should it become necessary in a clinical setting to suppress immune responses to allogeneic cell lines, we note that in a murine model of metastatic neuroblastoma, the immu-nosuppressive agent cyclosporin did not affect the tumor-tropic properties of the HB1.F3 cells (MK Danks
et al. unpublished observations). Whether the
immuno-genicity of cells from allogeneic sources will limit their utility will ultimately need to be evaluated in clinical trials, but multiple types of data indicate that the immunogenicity of at least some of these cell lines is not prohibitive. Consideration must also be given to the potential immunogenicity of the therapeutic gene or vector used to introduce this gene into the delivery cell line. In particular, there are clearly instances in which genes from non-human sources provide a greater therapeutic benefit. If the immunogenic potential of the vector or the transgene prohibits clinical use, obvious alternative are to use ‘empty vectors’, to delete immuno-genic sequences not required for protein function, or to ‘humanize’ immunogenic domains of non-human coding sequences, as has been reported for the rabbit CE that efficiently activates the prodrug irinotecan.114 Until
conditions are identified that permit unlimited expan-sion of clonal populations of autologous primary cells engineered to express a therapeutic of interest, well-characterized cell lines are most likely to have clinical utility.
Methods/vectors for engineering cells
to express therapeutic transgenes
Stem and progenitor cells used as delivery vehicles must be engineered to express the therapeutic of interest. Most Cell-mediated anticancer therapy
KS Aboodyet al
745
frequently, the therapeutic ‘payload’ is a peptide or protein, with transient or stable expression mediated by transduction with viral constructs. The use of adenovirus (replication-competent, -deficient or -selective), retro-virus or regulatable retroretro-virus,115,116 adeno-associated
virus,117HSV-1118 and HIV type 1119has been reported.
While a discussion of the advantages and disadvantages of each type of virus is beyond the scope of this review, major differences among these vectors include whether or not they integrate into the genome of the recipient cell, their ability to transduce the intended delivery cells, their immunogenic potential, and the level of transgene expression that can be expected from each. If long-term transgene expression is necessary, a vector that integrates into DNA such as retrovirus would be preferable; but then, as discussed above with respect to constructs used to generate immortalized cell lines as delivery vehicles, clonal populations of transduced cells must be isolated, expanded and the DNA integration sites mapped. Similarly, transfection methods could be used to introduce genes of interest into delivery cells, but this approach may also necessitate cloning and mapping of genomic integration sites. In contrast, adenoviral vectors can be used to mediate transient, high-level transgene expression without integration into the host cell genome, but then the immunogenic potential of intracellular adenovirus particles would need to be carefully evaluated. Circumvention or minimization of vector ‘issues’ will vary with each approach. Potentially, depending on the context of treatment, patients may already be immune deficient or the site of pathology might be in an immune-privileged site, thereby mini-mizing the potential for induction of an immune response.
Choice of therapeutic transgene
Stem and progenitor cells can be engineered to express various antitumor gene products, and deliver these products selectively to tumor foci. Gene products that have been evaluated thus far include prodrug-activating enzymes, inducers of apoptosis, differentiating agents, cell cycle modulators, anti-angiogenesis factors, im-mune-enhancing agents and oncolytic factors (Table 1). Cellular vehicles may also serve as virus producer cells, allowing more protracted and selective delivery of oncolytic viruses.120 Different types of therapeutics will
likely be best delivered by different types of cells that have been engineered specifically to target a given disease. For most anticancer applications, it seems reasonable transient high-level expression of therapeutic entities would produce the most robust and durable responses. Appropriate (effective) vector design and ‘therapeutic entities’ will depend heavily on the tumor targeted. While an extensive review of transgenes is beyond the scope of this commentary, a representative list of genes and tumor types that have been investigated is found in Table 1. In each case, the preclinical outcome likely depended on the level and duration of expression of the therapeutic gene at tumor foci, the dosing schedule of cell-mediated and -associated treatments, the appro-priateness of the molecular target, and the ability of the therapeutic entities to interact with and modify the function of that target.
Therapeutic efficacy of tumor-selective
cell-mediated gene delivery in preclinical
models
General considerations
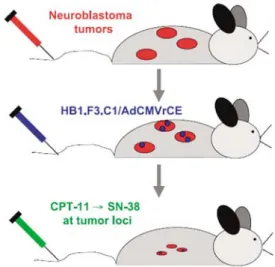
The majority of studies thus far have used MSCs or NSCs engineered to express an enzyme that activates a nontoxic prodrug. The hypothesis evaluated by this approach is based on the expression of the prodrug-activating enzyme preferentially at tumor foci, and the production of high concentrations of active drug at tumor sites. A specific example of this approach is shown in Figure 2, however, similar approaches using neural stem/progenitor cells to deliver various therapeutic genes have been used for other solid tumors, including glioma, medulloblastoma, melanoma, breast and pros-tate cancer (Table 1).
It is envisioned that this type of approach will improve therapeutic efficacy without additional toxicity. Essential to the anticipated improvement in therapeutic index is not only an increase of active drug at tumor foci, but also demonstration that the plasma concentration of active drug or therapeutic transgene does not increase com-pared to treatment with prodrug alone. Two recent publications examined these parameters. The first evalu-ated levels of enzyme and activevalu-ated prodrug in systemic circulation of mice treated with HB1.F3 cells expressing a CE that efficiently converts the prodrug CPT-11 to SN-38.16 Plasma levels of CE activity and SN-38 were
comparable in mice receiving HB1.F3 cells transduced with adenovirus to express CE and CPT-11 compared to CPT-11 alone. The second study quantitated plasma levels of interferon-b in mice treated with HB1.F3 cells expressing this cytokine. Circulating levels of IFN-bwere undetectable in mice receiving 2 million HB1.F3 cells
Figure 2 Schematic representation of neural stem/progenitor cell-directed enzyme pro-drug therapy (NDEPT). Mice bearing disseminated neuroblastoma receive i.v. injections of HB1.F3 cells transduced to express a secreted form of rCE. The NSPCs migrate selectively to tumorsin vivoand secrete rCE at tumor sites, which converts systemically administered CPT-11 to its active form, SN-38 a potent topoisomerase I inhibitor. High concentrations of SN-38 at tumor loci are cytotoxic to tumors, without additional toxic side effects. Based on Figure 1 from Dankset al.,16with permission of the American Association for Cancer Research.
transduced with adenovirus to express IFN-b.17 These
results suggest that little ‘leakage’ away from tumor foci occurs and that the probability is good that tumor-selective therapy can be achieved using this approach (Figure 3).
Specific considerations
For additional information regarding specific genes, we refer the reader to recent reviews that focus on prodrug-activating enzymes used in gene therapy.67,121,122 In the
brief discussions that follow, our intent is to highlight notable specific or unique features of research design or preclinical therapeutic outcomes in published studies focused on individual therapeutic transgenes.
Cytosine deaminase
E. coli CD (EC3.5.4.1) converts 5-FC to 5-fluorouridine
(5-FU). 5-FU, a pyrimidine analogue, is metabolized
in vivo by mammalian enzymes to active forms that
inhibit DNA and RNA synthesis. We have used HB1.F3 cells transduced to express CD. This CD-expressing cell line was generated using an amphotropic replication-deficient retrovirus in which CD expression is regulated by the retrovirus LTR. A clonal cell population was isolated following selection of transduced cells with puromycin. The established clonal cell line expresses functional CD. Single copies of the therapeutic gene (CD) and of the immortalizing gene (v-myc) inserted into ‘relatively barren’ regions of the host cell genome (KS Aboody and MK Danks, unpublished observations). The karyotype of the isolated cell line is stable for at least 26 passagesin vitro. Systemically administered HB1.F3 cells and also 5-FC cross the blood-brain barrier, making this approach uniquely well suited for the treatment of CNS tumors. Further, CD/5-FC produces a significant by-stander effect.123Using this approach, we have observed
470–80% decrease in tumor volume of mice bearing orthotopic invasive gliomas or intracranial melanoma, compared to the tumor volume in untreated mice or mice receiving NSCs or prodrug only.3,10The CD/5-FC
strategy also merits evaluation in eradicating or prevent-ing liver metastases of colon adenocarcinoma, since 5-FU is front-line for the treatment of this tumor type.
Carboxylesterase
Another enzyme/prodrug combination that shows pro-mise in preclinical studies is that of rabbit CE/CPT-11 (irinotecan).15,16,124,125Studies from our laboratory using
this combination employed HB1.F3 cells transduced with adenovirus to express a rabbit CE (rCE; EC.3.1.1.1) that efficiently converts CPT-11 to SN-38, a potent topoisome-rase I inhibitor. Expression of rCE is regulated by the CMV promoter. Intravenous administration of HB1.F3.rCE cells and CPT-11 produced long-term survi-val (greater than one year) in 90% of mice bearing multiple disseminated (metastatic) neuroblastoma. To our knowledge, this is the most durable and complete response published thus far using cell-mediated delivery approaches. The durable response may be attributable to the optimized ‘matching’ of the cells, vector, enzyme, prodrug and schedules of administration of each component with the targeted tumor type. Adenovirus produced extremely high levels of rCE expression. rCE activates CPT-11 more efficiently than human enzymes. CPT-11 has shown encouraging, albeit not curative,
activity in neuroblastoma patients. Efficacious schedules of administration of CPT-11 for the treatment of neuroblastoma are known.126 Each component of the
‘system’ was targeted specifically to try to attain complete remissions without recurrence specifically for disseminated neuroblastoma tumors.16As is the case for
CD/5FC, the combination of CE/CPT-11 also merits evaluation for potential efficacy in the treatment of colon adenocarcinoma, since CPT-11 is one of the most effective agents for the treatment of this tumor type.
Other enzyme/prodrug combinations
The two enzyme/prodrug combinations discussed above represent a small portion of combinations that have been reported in the literature; and the reader is referred to the previously mentioned excellent reviews for additional information on this topic. However, we propose that the efficacy of the activated prodrug in treating human tumors should be a primary consideration of which prodrugs are investigated in gene therapy enzyme/ prodrug studies in the future. If, clinically, antitumor efficacy of a given activated prodrug has relatively limited activity in the treatment of human tumors, then time, intellectual and financial resources would be better spent designing and characterizing novel combinations rather than conducting additional proof of principle experiments with available combinations simply because they are available.
Interleukins
Using a different therapeutic approach, SV40 large T antigen-immortalized primary mouse NSCs transduced with retrovirus to express interleukin 4 (IL-4) have been evaluated for efficacy in treating C6 rat gliomas or syngeneic GL261 mouse gliomas.2 IL-4 enhances
T-cell-mediated immune responses to tumor cells.127Treatment
of IL-4-expressing NSCs produced a 90-day survival of
480% of mice when tumor cells were co-injected with IL-4-secreting NSCs, compared to 0% 30-day survival in untreated mice. When a similar approach was used to Figure 3 Kaplan–Meier plots of tumor-bearing mice treated with HB1.F3/AdCMVrCE and CPT-11. Mice (10 per group) were injected i.v. with 500 000 SK-N-AS cells to produce disseminated neuro-blastoma (NB) tumors in 100% of mice. Two weeks later, mice were treated with rCE/CPT-11 NDEPT using the following treatment protocol: week 1, 2 million HB1.F3.C1 cells transduced with a MOI of 20 of AdCMVrCE followed by CPT-11 daily five times i.v. at the indicated doses; week 2, repeat of week 1; weeks 3 and 4, no treatment and weeks 5 and 6, same as weeks 1 and 2. The dose of CPT-11 administered was 15 mg kg 1. Based on Figure 3 from Dankset al.,16 with permission of the American Association for Cancer Research.
Cell-mediated anticancer therapy KS Aboodyet al
747
treat established GL261 tumors, 71% of mice survived for 90 days, whereas only 33% of tumor-bearing mice that received control NSCs were alive at this timepoint.2In a
similar study, immortalized Sprague–Dawley embryonic rat NSCs transduced with retrovirus to express IL-4 were implanted into C6 gliomas, which resulted in prolonged survival of the treated rats, when compared to untreated controls.2 In other studies, treatment of GL261
tumor-bearing rats with NSC-IL-12 cells significantly prolonged survival and produced long-term antitumor immunity compared to animals treated with unmodified NSCs.4,128
Bone marrow-derived neural stem-like cells (BM-NSCs) have also been used to express and deliver another interleukin: interleukin-23 (IL-23). Administra-tion of these BM-NSC-IL-23 cells inhibited tumor growth rate in C57BL/6 mice bearing gliomas.9 CD8+ T-cells
were essential for producing the observed antitumor effects mediated by IL-23-expressing BM-NSCs. Impor-tantly, the IL-23-expressing BM-NSC-treated mice that survived were resistant to tumor re-challenge. These data show that IL-23-expressing BM-NSCs induce immune-mediated antitumor effects in preclinical models of intracranial glioma.
Tumor necrosis factor-related apoptosis-inducing ligand (APO-2L)
NSCs transduced with an adenoviral vector encoding human tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) have also shown efficacy in rodent models of intracranial glioma.5 These modified NSCs
produced an increase in apoptotic cells in these tumors, and were associated with a significant inhibition in the rate of tumor progression. Importantly, Shah et al.7
designed a secreted version of TRAIL (S-TRAIL) to improve access of the therapeutic gene product to its molecular target, a cell membrane surface protein on cells adjacent to the NSCs. Intracranially implanted NSCs expressing S-TRAIL migrated into the gliomas and effected a 480% reduction in tumor growth. Since TRAIL induces apoptosis in tumor cells,129–131 but has
relatively few effects on most normal cells,132 TRAIL
is attractive as potentially useful in gene therapy approaches to treatment.
PEX
Another type of protein product that has been evaluated for efficacy in treating intracranial gliomas is the human metalloproteinase-2 fragment PEX. PEX inhibits glioma and endothelial cell proliferation and migration, and angiogenesis.133,134 The antitumor activity of human
HB1.F3 cells expressing PEX was investigated in mice bearing malignant gliomas. Intratumoral injections of these modified NSCs reduced tumor volume by 90%,8
and this reduction was concomitant with a decrease in angiogenesis and cell proliferation. Given the critical role of metalloproteinases in tumor invasion and progression, additional approaches to inhibit MMP function or, perhaps, to design prodrugs that are activated by MMPs, need to be investigated further.
Interferon-b
A fourth type of cell-mediated approach to the treatment of solid tumors has been reported by Dickson et al.17
These investigators used HB1.F3 cells to deliver
inter-feron-b to tumor foci, with the intent of normalizing tumor vasculature prior to administration of cytotoxic agents.135 The expectation was that by improving the
functional status of tumor vasculature, chemotherapeutic agents given subsequently would penetrate the tumor mass more extensively and a significant increase in their antitumor efficacy would be observed. HB1.F3 cells expressing interferon-b, when given alone, had little antitumor effect in mice bearing orthotopic neuroblasto-ma tumors; but these cells significantly enhanced the antitumor effect of cyclophosphamide, a chemothera-peutic agent used in the treatment of neuroblastoma. Studies are currently underway to evaluate the ability of HB1.F3.IFN-b cells to sensitize tumors to other chemo-therapeutic agents such as CPT-11 or doxorubicin that are relatively efficacious in the treatment of this tumor type. This general approach, with appropriate choice of chemotherapeutic agent, could be applicable to the treatment of other solid tumors as well.
Summary and future directions
The poor prognosis for patients with metastatic and invasive tumors and the toxic side effects of currently available treatments necessitate the development of effective tumor-selective therapies. Current data suggest that the inherent tumor-tropic properties of stem/ progenitor cells can be exploited to develop safe effective targeted therapy for these cancers. Several candidate genes/gene products have been mentioned in this commentary. However, more importantly, emerging safety and efficacy data for cell-mediated delivery will hopefully stimulate new efforts to identify novel genes for the treatment of specific tumor types. Further, studies to elucidate chemotactic factors and signaling pathways that regulate the tumor-tropism of stem and progenitor cells will facilitate identification of tumor types that would best respond to cell-mediated gene delivery.
In vivo human safety and efficacy data critical to the
success of the described approach include: cell distribu-tion and duradistribu-tion of survival, with and without admin-istration of conventional chemotherapy; identification of ‘safe’ genomic insertion sites for exogenous genes; confirmation of the predicted minimal immunogenic potential of stem/progenitor and cells; and minimization of the immunogenicity of non-human gene products. Further, and a theme throughout this commentary, it is likely that the type of delivery cell, the therapeutic gene and the vector used to engineer gene expression must be tailored to specific tumor types, if the optimal therapeu-tic benefit is to be realized. We also propose that, at least initially, cell-mediated approaches to cancer therapy will have the greatest impact in eradicating minimum residual disease in patients that have achieved apparent complete or near-complete remission with conventional therapy, but whose prognostic factors indicate that relapse with metastatic disease is highly likely.
Acknowledgements
This work was supported by National Institutes of Health/National Cancer Institute (CA113446, CA79763, CA76202, and CA21765); Stop Cancer Foundation, The
Rosalinde and Arthur Gilbert Foundation, Neidorf Family Foundation, HL Snyder Foundation, Jeanne Schnit-zer-Reynolds Foundation, Joseph Drown Foundation, and American Lebanese Syrian Associated Charities (ALSAC).
References
1 Herrlinger U, Woiciechowski C, Sena-Esteves M, Aboody KS, Jacobs AH, Rainov NGet al.Neural precursor cells for delivery of replication-conditional HSV-1 vectors to intracerebral gliomas.Mol Ther2000;1: 347–357.
2 Benedetti S, Pirola B, Pollo B, Magrassi L, Bruzzone MG, Rigamonti Det al.Gene therapy of experimental brain tumors using neural progenitor cells.Nat Med2000;6: 447–450. 3 Aboody KS, Brown A, Rainov NG, Bower KA, Liu S, Yang Wet
al.Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas.Proc Natl Acad Sci USA2000;97: 12846–12851.
4 Ehtesham M, Kabos P, Kabosova A, Neuman T, Black KL, Yu JS. The use of interleukin 12-secreting neural stem cells for the treatment of intracranial glioma.Cancer Res2002;62: 5657–5663. 5 Ehtesham M, Kabos P, Gutierrez MA, Chung NH, Griffith TS, Black KLet al.Induction of glioblastoma apoptosis using neural stem cell-mediated delivery of tumor necrosis factor-related apoptosis-inducing ligand.Cancer Res2002;62: 7170–7174. 6 Barresi V, Belluardo N, Sipione S, Mudo G, Cattaneo E,
Condorelli DF. Transplantation of prodrug-converting neural progenitor cells for brain tumor therapy.Cancer Gene Ther2003;
10: 396–402.
7 Shah K, Tung CH, Breakefield XO, Weissleder R. In vivo imaging of S-TRAIL-mediated tumor regression and apoptosis. Mol Ther2005;11: 926–931.
8 Kim SK, Cargioli TG, Machluf M, Yang W, Sun Y, Al-Hashem R et al.PEX-producing human neural stem cells inhibit tumor growth in a mouse glioma model. Clin Cancer Res 2005;11: 5965–5970.
9 Yuan X, Hu J, Belladonna ML, Black KL, Yu JS. Interleukin-23-expressing bone marrow-derived neural stem-like cells exhibit antitumor activity against intracranial glioma.Cancer Res2006;
66: 2630–2638.
10 Aboody KS, Najbauer J, Schmidt NO, Yang W, Wu JK, Zhuge Y et al.Targeting of melanoma brain metastases using engineered neural stem/progenitor cells.Neuro Oncol2006;8: 119–126. 11 Lin D, Najbauer J, Salvaterra PM, Mamelak AN, Barish ME,
Garcia Eet al.Novel method for visualizing and modeling the spatial distribution of neural stem cells within intracranial glioma.Neuroimage2007;37: S18–S26.
12 Li S, Gao Y, Tokuyama T, Yamamoto J, Yokota N, Yamamoto S et al. Genetically engineered neural stem cells migrate and suppress glioma cell growth at distant intracranial sites.Cancer Lett2007;251: 220–227.
13 Kim SK, Kim SU, Park IH, Bang JH, Aboody KS, Wang KCet al. Human neural stem cells target experimental intracranial medulloblastoma and deliver a therapeutic gene leading to tumor regression.Clin Cancer Res2006;12: 5550–5556. 14 Shimato S, Natsume A, Takeuchi H, Wakabayashi T, Fujii M, Ito
Met al.Human neural stem cells target and deliver therapeutic gene to experimental leptomeningeal medulloblastoma.Gene Therapy2007;14: 1132–1142.
15 Aboody KS, Bush RA, Garcia E, Metz MZ, Najbauer J, Justus KAet al.Development of a tumor-selective approach to treat metastatic cancer.PLoS ONE2006;1: e23.
16 Danks MK, Yoon KJ, Bush RA, Remack JS, Wierdl M, Tsurkan L et al.Tumor-targeted enzyme/prodrug therapy mediates long-term disease-free survival of mice bearing disseminated neuroblastoma.Cancer Res2007;67: 22–25.
17 Dickson PV, Hamner JB, Burger RA, Garcia E, Ouma AA, Kim SUet al.Intravascular administration of tumor tropic neural progenitor cells permits targeted delivery of interferon-beta and restricts tumor growth in a murine model of disseminated neuroblastoma.J Pediatr Surg2007;42: 48–53.
18 Nakamura K, Ito Y, Kawano Y, Kurozumi K, Kobune M, Tsuda Het al.Antitumor effect of genetically engineered mesenchy-mal stem cells in a rat glioma model.Gene Therapy 2004;11: 1155–1164.
19 Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J et al.Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas.Cancer Res2005;65: 3307–3318. 20 Miletic H, Fischer Y, Litwak S, Giroglou T, Waerzeggers Y,
Winkeler Aet al.Bystander killing of malignant glioma by bone marrow-derived tumor-infiltrating progenitor cells expressing a suicide gene.Mol Ther2007;15: 1373–1381.
21 Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res 2002;62: 3603–3608.
22 Elzaouk L, Moelling K, Pavlovic J. Anti-tumor activity of mesenchymal stem cells producing IL-12 in a mouse melanoma model.Exp Dermatol2006;15: 865–874.
23 Potapova I, Plotnikov A, Lu Z, Danilo Jr P, Valiunas V, Qu Jet al. Human mesenchymal stem cells as a gene delivery system to create cardiac pacemakers.Circ Res2004;94: 952–959. 24 Ehtesham M, Stevenson CB, Thompson RC. Stem cell therapies
for malignant glioma.Neurosurg Focus2005;19: E5.
25 Klein SM, Behrstock S, McHugh J, Hoffmann K, Wallace K, Suzuki Met al.GDNF delivery using human neural progenitor cells in a rat model of ALS.Hum Gene Ther2005;16: 509–521. 26 Hall B, Dembinski J, Sasser AK, Studeny M, Andreeff M,
Marini F. Mesenchymal stem cells in cancer: tumor-associated fibroblasts and cell-based delivery vehicles.Int J Hematol2007;
86: 8–16.
27 Kanehira M, Xin H, Hoshino K, Maemondo M, Mizuguchi H, Hayakawa Tet al.Targeted delivery of NK4 to multiple lung tumors by bone marrow-derived mesenchymal stem cells. Cancer Gene Ther2007;14: 894–903.
28 Kucerova L, Altanerova V, Matuskova M, Tyciakova S, Altaner C. Adipose tissue-derived human mesenchymal stem cells mediated prodrug cancer gene therapy. Cancer Res 2007;67: 6304–6313.
29 Mapara KY, Stevenson CB, Thompson RC, Ehtesham M. Stem cells as vehicles for the treatment of brain cancer.Neurosurg Clin N Am2007;18: 71–80, ix.
30 Najbauer J, Danks MK, Schmidt NO, Kim SU, Aboody KS. Neural stem cell-mediated therapy of primary and metastatic solid tumors. In: Bertolotti R, Ozawa K (eds).Progress in Gene Therapy, Autologous and Cancer Stem Cell Gene Therapy. World Scientific: Singapore, 2007.
31 Xin H, Kanehira M, Mizuguchi H, Hayakawa T, Kikuchi T, Nukiwa T et al. Targeted delivery of CX3CL1 to multiple lung tumors by mesenchymal stem cells.Stem Cells 2007;25: 1618–1626.
32 Ulrich AS. Biophysical aspects of using liposomes as delivery vehicles.Biosci Rep2002;22: 129–150.
33 Lee J, Elkahloun AG, Messina SA, Ferrari N, Xi D, Smith CL et al.Cellular and genetic characterization of human adult bone marrow-derived neural stem-like cells: a potential antiglioma cellular vector.Cancer Res2003;63: 8877–8889.
34 Miller AD. Nonviral liposomes. Methods Mol Med 2004; 90: 107–137.
35 Merdan T, Kopecek J, Kissel T. Prospects for cationic polymers in gene and oligonucleotide therapy against cancer.Adv Drug Deliv Rev2002;54: 715–758.
36 Brokx R, Gariepy J. Peptide- and polymer-based gene delivery vehicles.Methods Mol Med2004;90: 139–160.
Cell-mediated anticancer therapy KS Aboodyet al
749
37 Bharali DJ, Klejbor I, Stachowiak EK, Dutta P, Roy I, Kaur Net al.Organically modified silica nanoparticles: a nonviral vector forin vivogene delivery and expression in the brain.Proc Natl Acad Sci USA2005;102: 11539–11544.
38 Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy.Nat Rev Genet2007;8: 573–587. 39 Wagner E, Zatloukal K, Cotten M, Kirlappos H, Mechtler K,
Curiel DT et al. Coupling of adenovirus to transferrin-polylysine/DNA complexes greatly enhances receptor-mediated gene delivery and expression of transfected genes. Proc Natl Acad Sci USA1992;89: 6099–6103.
40 Joo SY, Kim JS. Enhancement of gene transfer to cervical cancer cells using transferrin-conjugated liposome. Drug Dev Ind Pharm2002;28: 1023–1031.
41 Zhu ZB, Makhija SK, Lu B, Wang M, Rivera AA, Preuss Met al. Transport across a polarized monolayer of Caco-2 cells by transferrin receptor-mediated adenovirus transcytosis.Virology 2004;325: 116–128.
42 Daniels TR, Ng PP, Delgado T, Lynch MR, Schiller G, Helguera Get al.Conjugation of an anti transferrin receptor IgG3-avidin fusion protein with biotinylated saporin results in significant enhancement of its cytotoxicity against malignant hemato-poietic cells.Mol Cancer Ther2007;6: 2995–3008.
43 Peng JL, Wu S, Zhao XP, Wang M, Li WH, Shen X et al. Downregulation of transferrin receptor surface expression by intracellular antibody.Biochem Biophys Res Commun2007;354: 864–871.
44 Tang Y, Han T, Everts M, Zhu ZB, Gillespie GY, Curiel DTet al. Directing adenovirus across the blood-brain barrier via melanotransferrin (P97) transcytosis pathway in an in vitro model.Gene Therapy2007;14: 523–532.
45 Xia CF, Zhang Y, Zhang Y, Boado RJ, Pardridge WM. Intravenous siRNA of brain cancer with receptor targeting and avidin-biotin technology.Pharm Res2007;24: 2309–2316. 46 Prutki M, Poljak-Blazi M, Jakopovic M, Tomas D, Stipancic I,
Zarkovic N. Altered iron metabolism, transferrin receptor 1 and ferritin in patients with colon cancer.Cancer Lett2006;238: 188–196.
47 Kelter G, Steinbach D, Konkimalla VB, Tahara T, Taketani S, Fiebig HH et al. Role of transferrin receptor and the ABC transporters ABCB6 and ABCB7 for resistance and differentia-tion of tumor cells towards artesunate.PLoS ONE2007;2: e798. 48 Imitola J, Raddassi K, Park KI, Mueller FJ, Nieto M, Teng YDet al.Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci USA 2004; 101: 18117–18122.
49 Erlandsson A, Larsson J, Forsberg-Nilsson K. Stem cell factor is a chemoattractant and a survival factor for CNS stem cells.Exp Cell Res2004;301: 201–210.
50 Sun L, Lee J, Fine HA. Neuronally expressed stem cell factor induces neural stem cell migration to areas of brain injury. J Clin Invest2004;113: 1364–1374.
51 Heese O, Disko A, Zirkel D, Westphal M, Lamszus K. Neural stem cell migration toward gliomasin vitro.Neuro Oncol2005;7: 476–484.
52 Schmidt NO, Przylecki W, Yang W, Ziu M, Teng Y, Kim SUet al. Brain tumor tropism of transplanted human neural stem cells is induced by vascular endothelial growth factor.Neoplasia2005;
7: 623–629.
53 Widera D, Holtkamp W, Entschladen F, Niggemann B, Zanker K, Kaltschmidt Bet al.MCP-1 induces migration of adult neural stem cells.Eur J Cell Biol2004;83: 381–387.
54 Palumbo R, Bianchi ME. High mobility group box 1 protein, a cue for stem cell recruitment. Biochem Pharmacol 2004; 68: 1165–1170.
55 Palumbo R, Galvez BG, Pusterla T, De Marchis F, Cossu G, Marcu KBet al.Cells migrating to sites of tissue damage in
response to the danger signal HMGB1 require NF-kappaB activation.J Cell Biol2007;179: 33–40.
56 Rafii S, Lyden D. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat Med2003;9: 702–712.
57 Wysoczynski M, Reca R, Ratajczak J, Kucia M, Shirvaikar N, Honczarenko Met al.Incorporation of CXCR4 into membrane lipid rafts primes homing-related responses of hematopoietic stem/progenitor cells to an SDF-1 gradient.Blood 2005; 105: 40–48.
58 Son BR, Marquez-Curtis LA, Kucia M, Wysoczynski M, Turner AR, Ratajczak Jet al.Migration of bone marrow and cord blood mesenchymal stem cellsin vitrois regulated by stromal-derived factor-1-CXCR4 and hepatocyte growth factor-c-met axes and involves matrix metalloproteinases. Stem Cells2006; 24: 1254–1264.
59 Ziu M, Schmidt NO, Cargioli TG, Aboody KS, Black PM, Carroll RS. Glioma-produced extracellular matrix influences brain tumor tropism of human neural stem cells.J Neurooncol 2006;79: 125–133.
60 Ries C, Egea V, Karow M, Kolb H, Jochum M, Neth P. MMP-2, MT1-MMP, and TIMP-2 are essential for the invasive capacity of human mesenchymal stem cells: differential regulation by inflammatory cytokines.Blood2007;109: 4055–4063.
61 Aghi M, Hochberg F, Breakefield XO. Prodrug activation enzymes in cancer gene therapy.J Gene Med2000;2: 148–164. 62 Davidson BL, Breakefield XO. Viral vectors for gene delivery to
the nervous system.Nat Rev Neurosci2003;4: 353–364. 63 Rooseboom M, Commandeur JN, Vermeulen NP.
Enzyme-catalyzed activation of anticancer prodrugs. Pharmacol Rev 2004;56: 53–102.
64 Fischer U, Steffens S, Frank S, Rainov NG, Schulze-Osthoff K, Kramm CM. Mechanisms of thymidine kinase/ganciclovir and cytosine deaminase/5-fluorocytosine suicide gene therapy-induced cell death in glioma cells.Oncogene2005;24: 1231–1243.
65 Lee KC, Hamstra DA, Bullarayasamudram S, Bhojani MS, Moffat BA, Dornfeld KJet al. Fusion of the HSV-1 tegument protein vp22 to cytosine deaminase confers enhanced bystan-der effect and increased therapeutic benefit.Gene Therapy2006;
13: 127–137.
66 Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond.Nat Rev Cancer2006;6: 789–802.
67 Shah K, Breakefield XO. HSV amplicon vectors for cancer therapy.Curr Gene Ther2006;6: 361–370.
68 Guffey MB, Parker JN, Luckett Jr WS, Gillespie GY, Meleth S, Whitley RJet al.Engineered herpes simplex virus expressing bacterial cytosine deaminase for experimental therapy of brain tumors.Cancer Gene Ther2007;14: 45–56.
69 Jeong SW, Chu K, Jung KH, Kim SU, Kim M, Roh JK. Human neural stem cell transplantation promotes functional recovery in rats with experimental intracerebral hemorrhage.Stroke2003;
34: 2258–2263.
70 Kim SU. Human neural stem cells genetically modified for brain repair in neurological disorders.Neuropathology2004;24: 159–171.
71 Kim SU, Park IH, Kim TH, Kim KS, Choi HB, Hong SHet al. Brain transplantation of human neural stem cells transduced with tyrosine hydroxylase and GTP cyclohydrolase 1 provides functional improvement in animal models of Parkinson disease.Neuropathology2006;26: 129–140.
72 Kim SU. Genetically engineered human neural stem cells for brain repair in neurological diseases. Brain Dev2007;29: 193–201.
73 Lee HJ, Kim KS, Park IH, Kim SU. Human neural stem cells over-expressing VEGF provide neuroprotection, angiogenesis and functional recovery in mouse stroke model. PLoS ONE 2007;2: e156.
74 Dittmar T, Seidel J, Zaenker KS, Niggemann B. Carcinogenesis driven by bone marrow-derived stem cells.Contrib Microbiol 2006;13: 156–169.
75 Chen EH, Grote E, Mohler W, Vignery A. Cell-cell fusion.FEBS Lett2007;581: 2181–2193.
76 Duelli D, Lazebnik Y. Cell-to-cell fusion as a link between viruses and cancer.Nat Rev Cancer2007;7: 968–976.
77 Studeny M, Marini FC, Dembinski JL, Zompetta C, Cabreira-Hansen M, Bekele BNet al.Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents.J Natl Cancer Inst2004;96: 1593–1603. 78 Hall B, Andreeff M, Marini F. The participation of
mesenchy-mal stem cells in tumor stroma formation and their application as targeted-gene delivery vehicles.Handb Exp Pharmacol2007;
180: 263–283.
79 Stoff-Khalili MA, Rivera AA, Mathis JM, Banerjee NS, Moon AS, Hess A et al. Mesenchymal stem cells as a vehicle for targeted delivery of CRAds to lung metastases of breast carcinoma.Breast Cancer Res Treat2007;105: 157–167.
80 Takahashi K, Okita K, Nakagawa M, Yamanaka S. Induction of pluripotent stem cells from fibroblast cultures.Nat Protoc2007;
2: 3081–3089.
81 Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131: 861–872.
82 Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S et al. Induced pluripotent stem cell lines derived from human somatic cells.Science2007;318: 1917–1920. 83 Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts.Nat Biotechnol 2008;26: 101–106.
84 Nosrat IV, Smith CA, Mullally P, Olson L, Nosrat CA. Dental pulp cells provide neurotrophic support for dopaminergic neurons and differentiate into neuronsin vitro; implications for tissue engineering and repair in the nervous system. Eur J Neurosci2004;19: 2388–2398.
85 Chan J, O’Donoghue K, de la Fuente J, Roberts IA, Kumar S, Morgan JE et al. Human fetal mesenchymal stem cells as vehicles for gene delivery.Stem Cells2005;23: 93–102. 86 Wei J, Blum S, Unger M, Jarmy G, Lamparter M, Geishauser A
et al. Embryonic endothelial progenitor cells armed with a suicide gene target hypoxic lung metastases after intravenous delivery.Cancer Cell2004;5: 477–488.
87 Kern S, Eichler H, Stoeve J, Kluter H, Bieback K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells 2006; 24: 1294–1301.
88 Pike-Overzet K, van der Burg M, Wagemaker G, van Dongen JJ, Staal FJ. New insights and unresolved issues regarding insertional mutagenesis in X-linked SCID gene therapy. Mol Ther2007;15: 1910–1916.
89 Cacci E, Villa A, Parmar M, Cavallaro M, Mandahl N, Lindvall O et al. Generation of human cortical neurons from a new immortal fetal neural stem cell line. Exp Cell Res 2007;
313: 588–601.
90 De Filippis L, Lamorte G, Snyder EY, Malgaroli A, Vescovi AL. A novel, immortal, and multipotent human neural stem cell line generating functional neurons and oligodendrocytes.Stem Cells2007;25: 2312–2321.
91 Lee HJ, Kim KS, Kim EJ, Choi HB, Lee KH, Park IHet al.Brain transplantation of immortalized human neural stem cells promotes functional recovery in mouse intracerebral hemor-rhage stroke model.Stem Cells2007;25: 1204–1212.
92 Bai Y, Hu Q, Li X, Wang Y, Lin C, Shen Let al.Telomerase immortalization of human neural progenitor cells.Neuroreport 2004;15: 245–249.
93 Xu C, Jiang J, Sottile V, McWhir J, Lebkowski J, Carpenter MK. Immortalized fibroblast-like cells derived from human em-bryonic stem cells support undifferentiated cell growth. Stem Cells2004;22: 972–980.
94 Natesan S. Telomerase extends a helping hand to progenitor cells.Trends Biotechnol2005;23: 1–3.
95 Terai M, Uyama T, Sugiki T, Li XK, Umezawa A, Kiyono T. Immortalization of human fetal cells: the life span of umbilical cord blood-derived cells can be prolonged without manipulat-ing p16INK4a/RB brakmanipulat-ing pathway. Mol Biol Cell 2005; 16: 1491–1499.
96 Honma T, Honmou O, Iihoshi S, Harada K, Houkin K, Hamada Het al.Intravenous infusion of immortalized human mesench-ymal stem cells protects against injury in a cerebral ischemia model in adult rat.Exp Neurol2006;199: 56–66.
97 Zhang X, Soda Y, Takahashi K, Bai Y, Mitsuru A, Igura Ket al. Successful immortalization of mesenchymal progenitor cells derived from human placenta and the differentiation abilities of immortalized cells.Biochem Biophys Res Commun2006;351: 853–859.
98 Huang Q, Chen M, Liang S, Acha V, Liu D, Yuan F et al. Improving cell therapy—experiments using transplanted telo-merase-immortalized cells in immunodeficient mice. Mech Ageing Dev2007;128: 25–30.
99 Takeuchi M, Takeuchi K, Kohara A, Satoh M, Shioda S, Ozawa Yet al.Chromosomal instability in human mesenchymal stem cells immortalized with human papilloma virus E6, E7, and hTERT genes.In Vitro Cell Dev Biol Anim2007;43: 129–138. 100 Truckenmiller ME, Vawter MP, Zhang P, Conejero-Goldberg C,
Dillon-Carter O, Morales N et al. AF5, a CNS cell line immortalized with an N-terminal fragment of SV40 large T: growth, differentiation, genetic stability, and gene expression. Exp Neurol2002;175: 318–337.
101 Yeager TR, Reddel RR. Constructing immortalized human cell lines.Curr Opin Biotechnol1999;10: 465–469.
102 Serakinci N, Christensen R, Graakjaer J, Cairney CJ, Keith WN, Alsner J et al. Ectopically hTERT expressing adult human mesenchymal stem cells are less radiosensitive than their telomerase negative counterpart.Exp Cell Res2007;313: 1056–1067.
103 Richardson RM, Nguyen B, Holt SE, Broaddus WC, Fillmore HL. Ectopic telomerase expression inhibits neuronal differen-tiation of NT2 neural progenitor cells.Neurosci Lett2007;421: 168–172.
104 Foroni C, Galli R, Cipelletti B, Caumo A, Alberti S, Fiocco Ret al. Resilience to transformation and inherent genetic and functional stability of adult neural stem cells ex vivo.Cancer Res2007;67: 3725–3733.
105 Terada N, Hamazaki T, Oka M, Hoki M, Mastalerz DM, Nakano Y et al. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature 2002; 416: 542–545.
106 Ying QL, Nichols J, Evans EP, Smith AG. Changing potency by spontaneous fusion.Nature2002;416: 545–548.
107 Ianus A, Holz GG, Theise ND, Hussain MA.In vivoderivation of glucose-competent pancreatic endocrine cells from bone marrow without evidence of cell fusion.J Clin Invest2003;111: 843–850.
108 Wurmser AE, Nakashima K, Summers RG, Toni N, D’Amour KA, Lie DC et al. Cell fusion-independent differentiation of neural stem cells to the endothelial lineage. Nature2004;430: 350–356.
109 Brittan M, Braun KM, Reynolds LE, Conti FJ, Reynolds AR, Poulsom R et al. Bone marrow cells engraft within the epidermis and proliferate in vivo with no evidence of cell fusion.J Pathol2005;205: 1–13.
110 Sato Y, Araki H, Kato J, Nakamura K, Kawano Y, Kobune M et al.Human mesenchymal stem cells xenografted directly to Cell-mediated anticancer therapy
KS Aboodyet al
751