w ww . e l s e v i e r . c o m / l o c a t e / b j p
Original
article
Induction
of
apoptosis
by
Moutan
Cortex
Radicis
in
human
gastric
cancer
cells
through
the
activation
of
caspases
and
the
AMPK
signaling
pathway
Cheol
Park
a,
Min-Ho
Han
b,
Shin-Hyung
Park
c,
Su-Hyun
Hong
d,
Gi-Young
Kim
e,
Sung-Kwon
Moon
f,
Wun-Jae
Kim
g,∗,
Yung
Hyun
Choi
d,h,∗aDepartmentofMolecularBiology,CollegeofNaturalSciencesandHumanEcology,DongeuiUniversity,Busan,RepublicofKorea bNaturalProductsResearchTeam,MarineBiodiversityInstituteofKorea,Seocheon,RepublicofKorea
cDepartmentofPathology,DongeuiUniversityCollegeofKoreanMedicine,Busan,RepublicofKorea dDepartmentofBiochemistry,DongeuiUniversityCollegeofKoreanMedicine,Busan,RepublicofKorea
eDepartmentofMarineLifeSciences,SchoolofMarineBiomedicalScience,JejuNationalUniversity,Jeju,RepublicofKorea
fDepartmentofUrology,ChungbukNationalUniversity,CollegeofMedicineandInstituteforTumorResearch,Cheongju,RepublicofKorea gSchoolofFoodScienceandTechnology,Chung-AngUniversity,Ansung,RepublicofKorea
hAnti-AgingResearchCenter&Blue-BioIndustryRIC,DongeuiUniversity,Busan,RepublicofKorea
a
r
t
i
c
l
e
i
n
f
o
Articlehistory:
Received6September2016 Accepted18November2016 Availableonline24December2016
Keywords:
MoutanCortexRadicis
Gastriccancer Apoptosis Caspase AMPK
a
b
s
t
r
a
c
t
MoutanCortexRadicis,therootbarkofPaeonia×suffruticosaAndrews,Paeoniaceae,hasbeenwidely usedintraditionalmedicinetherapy.Althoughithasbeenshowntopossessmanypharmacological activities,themolecularmechanismsofitsanti-canceractivityhavenotbeenclearlyelucidated.Inthe presentstudy,weinvestigatedthepro-apoptoticeffectsoftheethanolextractofMoutanCortexRadicis
inhumangastriccancerAGScells.MoutanCortexRadicistreatmentinhibitedthecellviabilityofAGScells inaconcentration-dependentmanner,whichwasassociatedwithapoptoticcelldeath.MoutanCortex Radicis’sinductionofapoptosiswasconnectedwiththeupregulationofdeathreceptor4,deathreceptor 5,tumornecrosisfactor-relatedapoptosis-inducingligand,Fasligand,andBax,andthedownregulation ofBcl-2andBid.MoutanCortexRadicistreatmentalsoinducedthelossofmitochondrialmembrane potential( m),theproteolyticactivationofcaspases(-3,-8,and-9),andthedegradationof poly(ADP-ribose)polymerase,anactivatedcaspase-3substrateprotein.However,thepre-treatmentofacaspase-3 inhibitorsignificantlyattenuatedMoutanCortexRadicis-inducedapoptosisandcellviabilityreduction.In addition,MoutanCortexRadicistreatmenteffectivelyactivatedtheadenosinemonophosphate-activated proteinkinasesignalingpathway;however,aspecificinhibitorofAMPKsignificantlyreducedMoutan
CortexRadicis-inducedapoptosis.Overall,theresultssuggestthattheapoptoticactivityofMoutanCortex Radicismaybeassociatedwithacaspase-dependentcascadethroughtheactivationofbothextrinsic andintrinsicsignalingpathwaysconnectedwithadenosinemonophosphate-activatedproteinkinase activation,andMoutanCortexRadicisasanactivatorofadenosinemonophosphate-activatedprotein kinasecouldbeaprospectiveapplicationtotreathumancancers.
©2016SociedadeBrasileiradeFarmacognosia.PublishedbyElsevierEditoraLtda.Thisisanopen accessarticleundertheCCBY-NC-NDlicense(http://creativecommons.org/licenses/by-nc-nd/4.0/).
Introduction
Apoptosisisawell-knowntypeofprogrammedcelldeath medi-atedbytheextrinsicandintrinsicpathways.Theextrinsicpathway isactivatedbyaninteractionbetweendeathreceptors(DR)and theirligandsattheplasmamembrane,whichcausesthe subse-quentactivationofcaspase-8(FuldaandDebatin,2006;Kantari
∗ Correspondingauthors.
E-mails:[email protected](W.Kim),[email protected](Y.H.Choi).
andWalczak,2011).Theintrinsicpathway,termedthe mitochon-drialpathway,isinitiatedbythelossofmitochondrialmembrane potential(MMP, m)andthereleaseofpro-apoptoticproteins, includingcytochromec,leadingtotheactivationofcaspase-9and -3(MacKenzieandClark,2008;Hensleyetal.,2013).The extrin-sicpathwaycanalsocrosstalkwiththeintrinsicpathwaythrough thecaspase-8-mediatedtruncationofBid,amemberoftheBcl-2 familyofproteins,withanultimateamplificationoftheintrinsic apoptoticpathway(Lovelletal.,2008;Shamas-Dinetal.,2011).
AMP-activated proteinkinase(AMPK)is ametabolic-sensing protein kinase and plays a critical role as an energy-sensor in
http://dx.doi.org/10.1016/j.bjp.2016.11.003
ATP-deprived conditions (Gwinn et al., 2008; Shackelford and Shaw,2009;Tranetal.,2016).AnincreasedADP/ATPratioleads toAMPKphosphorylationatThr172,contributingtotheactivation oftheATP-generatingcatabolicprocesses,suchasfattyacid oxida-tionandglycolysis,aswellasthesuppressionoftheATP-utilizing anabolicpathways,includingglycogen,protein,andlipidsynthesis (Woodsetal.,2003;Shawetal.,2004).Recently,avarietyofstudies havereportedthatAMPKregulatescellproliferationandapoptosis viamultiplesignalingpathways,suchasphosphorylationandthe subsequentstabilizationoftumorsuppressorp53,the upregula-tionofcyclin-dependentkinaseinhibitors,andthedownregulation ofthemammaliantargetofrapamycincomplex-1activity(Gwinn etal.,2008; Monteverdeetal.,2015).In particular,ithasbeen demonstrated that AMPKactivation is responsible for inducing apoptosisviathehmitochondrialpathway(Nieminenetal.,2013; Fiarolaetal.,2015;Chenetal.,2015).Ontheotherhand,AMPKis alsoknowntoplayaprotectiveroleincancercellsundermetabolic stressedconditionsviathemaintenanceofenergyhomeostasisand modulationofautophagy,makingitsinfluenceoncancer progres-sioncontroversial(Laderouteetal.,2006;Rubinszteinetal.,2007; Jeon,2016).
MoutanCortexRadicis,the rootbarkof Paeonia×suffruticosa
Andrews,Paeoniaceae,hasbeenwidelyusedtoregulatehuman sickness, such as eliminating heat, promoting blood flow, and removingbloodstasisintraditionalmedicine(Hiraietal.,1983; Poonetal.,2011).Italsohasbeenreportedthattheextractsof MoutanCortexRadicisanditsphenoliccompoundsexhibit anti-oxidant(Rho etal.,2005), hepato-protective(Park et al.,2011), neuro-protective(Kimetal.,2014),anti-inflammatory(Fuetal., 2012;Yunet al.,2013),and anti-tyrosinase effects (Penget al., 2013).Morerecently,theanti-canceractivitiesofMoutanCortex Radicishavebeensuggested.Extractsandseveralcomponents iso-latedfromMCRwereproventoinducecellcyclearrestintheG1 phase(Gaoetal.,2015)andapoptosis(Choietal.,2012;Mukudai etal.,2013;Fanetal.,2013;Lietal.,2014;Gaoetal.,2015),inhibit invasion(Wangetal.,2012),and overcomedrugresistance(Cai etal.,2014)inavarietyofcancercelllines.However,theeffects ofMoutanCortexRadicisonAMPKsignaling-mediatedapoptosisin cancercellsandtheunderlyingdetailedmechanismshavenotbeen elucidatedtodate.Inthecurrentstudy,weexploredtheanti-cancer effectsofanethanolextractofMoutanCortexRadicis(MCR)inAGS humangastriccancercellsandinvestigatedtheunderlying mech-anism.WefoundthatMCRtriggeredcaspase-dependentapoptosis throughtheactivationofboththeintrinsicandextrinsicpathways, indicatingthatAMPKisacriticalregulatorofMCR-induced apo-ptosis.Toourknowledge,thisisthefirstpublicationtosuggestthe involvementofAMPKintheanti-canceractivityofMCR.
Materialsandmethods
Reagentsandantibodies
RPMI-1640andfetalbovineserum(FBS)wereobtainedfrom WelGENEInc.(Daegu,RepublicofKorea). 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT), 4,6-diamidino-2-phenylindole (DAPI), ethidium bromide (EtBr), 5,5′, 6,6′
-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide (JC-1),
and compound C, an inhibitor of AMPK, were purchased from Sigma–Aldrich Chemical Co. (St. Louis, MO, USA). An Annexin V-fluoresceinisothiocyanate(FITC)ApoptosisDetection Kit and a mitochondrial fractionation kit were purchased from Bec-ton Dickinson (San Jose, CA, USA) and Active Motif (Carlsbad, CA, USA), respectively. N -benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone (z-DEVD-fmk), a caspase-3 inhibitor, and caspasescolorimetricassaykitswereobtainedfromCalBiochem (San Diego, CA, USA) and R&D Systems (Minneapolis, MN,
USA),respectively.Theprimaryantibodieswerepurchasedfrom Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA) and Cell SignalingTechnology,Inc.(Beverly,MA,USA).Peroxidase-labeled donkey anti-rabbit and sheepanti-mouse immunoglobulin and an enhanced chemiluminescence (ECL) detection system were obtained from Amersham Co. (Arlington Heights, IL, USA). All otherchemicalsnotspecificallycitedherewerepurchasedfrom Sigma–AldrichChemicalCo.
PreparationofMCR
ToprepareMCR,thedried rootbarkofPaeonia×suffruticosa
Andrews,Paeoniaceae(100g),wasprovidedfromDongeuiKorean MedicalCenter(Busan,RepublicofKorea)andpulverizedintoa finepowder.Thepowderwasthenextractedin1lof70%ethanol bysonicationfor3h.Afterfilteringandconcentratingtheextracts, theremainedpowderwasdissolvedindimethylsulfoxide(DMSO) asa stock solutionat a100mg/mlconcentration andstored at 4◦C.Thestocksolutionwasdilutedwithmediumtothedesired
concentrationpriortouse.
Cellculture
AGS human gastric cancer and WI-38 human normal lung fibroblastcells wereobtainedfrom theAmerican Type Culture Collection (Manassas, MD, USA) and cultured in a RPMI-1640 mediumcontaining10% heat-inactivatedFBS and1% penicillin-streptomycinat37◦Cinahumidifiedatmospherecontaining5%
CO2.
Cellviabilityassay
Toinvestigatethecellviability,AGSandWI-38cellswereseeded ina96-wellplate(2×103cells/well)andstabilizedfor24h.The cells weretreated withvarious concentrationsof MCRfor 24h withorwithout1hofpre-treatmentofz-DEVD-fmkorcompound
C. AnMTT workingsolution(0.5mg/ml) wasthen addedtothe mediaandincubatedforfurther3hat37◦C.Afterincubation,the
culturesupernatantwasaspirated,and100lofDMSOwasadded
to completely dissolve the formazan crystals. The absorbance ofeachwellwasmeasuredatawavelengthof540nmusingan enzyme-linked immunosorbent assay(ELISA) reader(Molecular
Fig.3.TheeffectsofMoutanCortexRadicisontheexpressionlevelsofapoptosisregulatorsandMMPvaluesinAGScells.(AandB)AftertreatmentwithMCR,thecell lysateswereseparatedonSDS-polyacrylamidegelsandtransferredtoPVDFmembranes.Themembraneswereprobedwiththeindicatedantibodies,andtheproteinswere visualizedusinganECLdetectionsystem.Actinwasusedasaninternalcontrol.(C)ToevaluatethechangesinMMP,thecellswerestainedwithJC-1dyeandwerethen analyzedonaDNAflowcytometer.Thedataareexpressedasthemean±SDofthreeindependentexperiments(*p<0.05vs.untreatedcontrol).(D)Themitochondrial factions(M.F.)andcytosolicfactions(C.F.)wereseparatedbySDS-polyacrylamidegelelectrophoresis,followedbyWesternblotanalysisusinganti-cytochromecantibody. Cytochromecoxidasesubunit4(COXIV)andactinwereusedasinternalcontrolsforthemitochondrialandcytosolicfractions,respectively.
Devices,Sunnyvale,CA,USA)(Youetal.,2016).ToassessMCR’s effectoncellularmorphology,theAGScellswerephotographed underaninvertedmicroscope(CarlZeiss,Oberkochen,Germany).
NuclearstainingwithDAPI
AGS cells were treated with various concentrations of MCR for24hwithorwithout1hofpre-treatmentofz-DEVD-fmkor compoundC.Then,cellswereharvested,washedwith phosphate-bufferedsaline(PBS),and fixedwith3.7%paraformaldehydefor 30minatroomtemperature.Aftertheywerewashedtwicewith PBS,cellswereattachedonglassslidesusingcytospin(Shandon, Pittsburgh,PA,USA)andstainedwith2.5g/mlofDAPIsolutionfor
10minatroomtemperature.Thestainedcellswerewashedthree timeswithPBSandanalyzedusingafluorescencemicroscope(Carl Zeiss).
DNAfragmentationassay
FollowingMCRtreatment for 24hat various concentrations with or without 1h of pre-treatment of z-DEVD-fmk or com-pound C, the cells were lysed in a buffer [10mM of Tris–HCl (pHof7.4),150mMofNaCl,5mMofethylendiaminetetraacetic acid(EDTA), and 0.5% Triton X-100] for 1hat room tempera-ture.Aftercentrifugationat18,300×gfor30min,thesupernatant wascollectedandincubated withproteinaseK for3hat50◦C.
ThefragmentedDNAinsupernatantwaspurifiedusingthesame amountofneutralphenol:chloroform:isoamylalcoholsolutionby rotation for 30min at room temperature. After centrifugation, thesupernatantwasaddedwith0.5MofNaCl(final concentra-tion)and1volumeofisopropanoltoprecipitatethefragmented DNA and incubatedovernightat 4◦C. TheDNA pelletobtained
by centrifugation was then dissolved in TE buffer (10mM of Tris–HCl containing 1mM of EDTA) containing RNase A and separatedthrough 1%agarose gel.TheDNA fragmentation pat-ternwasvisualizedbyanultravioletlightsourcefollowingEtBr staining.
Flowcytometryanalysis
ChangesintheMMPweremeasuredbyflowcytometry (Bec-tonDickinson,SanJose,CA,USA) usingJC-1,thedual-emission potential-sensitiveprobe.Thecellstreatedwithvarious concen-trationsofMCRfor24hwerecollected,washedwithPBStwice, andincubatedwith10MofJC-1for20minat37◦C.inthedark.
Aftercentrifugation,thestainedcellswerewashedoncewithPBS toremoveunbounddyeandresuspendedwithPBS.Thereafter,the amountofJC-1retainedby10,000cellspersamplewasmeasured usingaflowcytometer.ForAnnexinV-propidiumiodide(PI)double staining,cellswerechallengedwithMCRfor24hwithorwithout 1hofpretreatmentofz-DEVD-fmkofcompoundC.Apoptoticcells werequantitativelyidentifiedwiththeAnnexinV-FITCApoptosis DetectionKitcontainingFITC-conjugatedAnnexinVandPI follow-ingtheprotocolsprovidedbythemanufacturer(Eometal.,2015). ThedatawereconvertedtodensityplotsusingCellQuestsoftware forpresentation.
Determinationofcaspaseactivity
Theactivitiesofcaspasesweredeterminedusingcolorimetric assay kits containing the synthetic tetrapeptides [Asp-Glu-Val-Asp(DEAD) forcaspase-3, Ile-Glu-Thr-Asp(IETD)for caspase-8,
and Leu-Glu- His-Asp (LEHD) for caspase-9] labeled with p -nitroaniline (pNA)accordingtothemanufacturer’sinstructions. Briefly, MCR-treated cells were harvested and lysed in the suppliedlysisbuffer.Supernatantswerethencollectedand incu-bated at 37◦C withthe supplied reaction buffer, dithiothreitol
(DTT), and respective substrates. Reactionactivities were eval-uated by measuring the absorbance at 405nm using an ELISA reader.
Westernblottinganalysis
To preparewhole celllysate, cells were lysed withice-cold lysisbuffer[25mMofTris–Cl(pHof7.5),250mMofNaCl,5mM ofEDTA,1%NonidetP-40,1mMofphenymethylsulfonylfluoride (PMSF),5mMofDTT]includingacompleteproteaseinhibitor cock-tailtabletandphosphataseinhibitors(1mMofNa3VO4,100mM ofNaF,10mMofNaPP).Inaparallelexperiment,the mitochon-drialandcytosolicfractionswereisolatedusingamitochondrial fractionation kit according to the manufacturer’s instructions. Aftercentrifugation at 15,800×g at 4◦C for 30min,the
super-natantswerecollected,andproteinconcentrationwasdetermined using a Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA,USA),accordingtothemanufacturer’sinstructions.Equivalent amountsofproteinwereresolvedbysodiumdodecylsulfate (SDS)-polyacrylamide gels and transferred to polyvinylidene fluoride (PVDF)membranes(Millipore,Bedford,MA,USA).Themembranes wereprobedwiththespecificprimaryantibodiesand correspond-ingsecondaryantibodies.Theprotein-antibodycomplexes were detectedbyanECLdetectionsystemaccordingtothe manufac-turer’sprotocol.
Statisticalanalysis
Eachresultisexpressedasthemean±standarddeviation(SD) ofdataobtainedfromindependenttriplicateexperiments.The sta-tisticalanalysiswasperformedusingapairedStudent’st-test.A
p-value<0.05wasconsideredstatisticallysignificant.
Results
MCRsuppressedcellproliferationbyinducingapoptosisinAGS cells
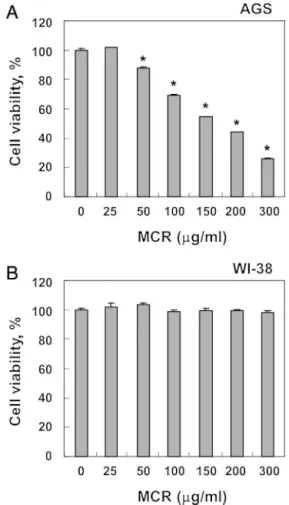
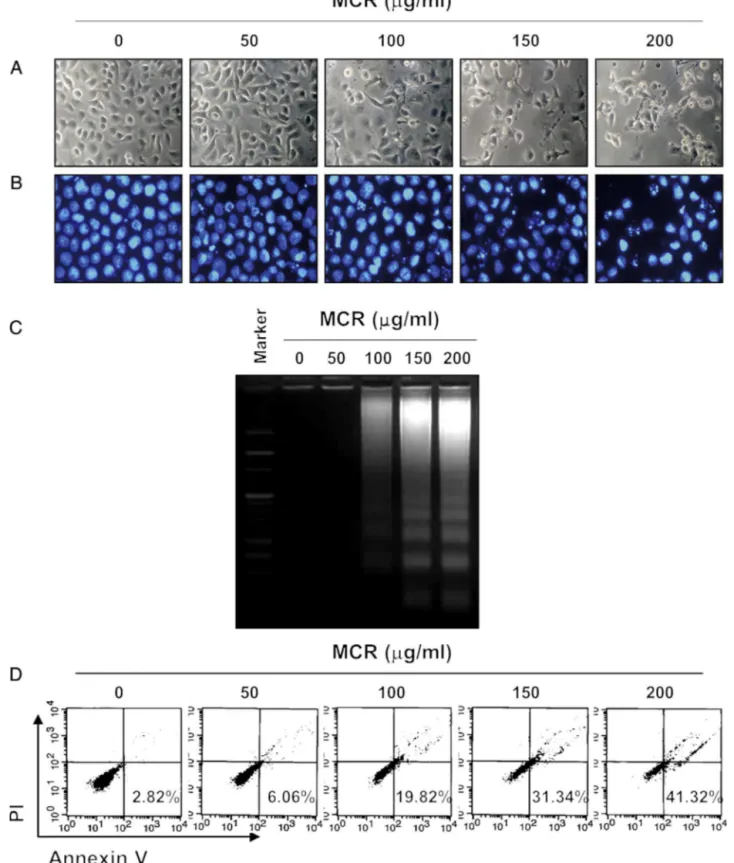
To investigate the effects of MCR on thecell viability,AGS cellsweretreated withvariousconcentrationsof MCRfor24h. AsshowninFig.1A,MCRtreatmentreducedthecellviabilityina concentration-dependentmanner.Morphologicalchanges, includ-ingmembraneblebbing,diminishedcelldensity,andanincreased numberof floatingcells,werealsoobserved(Fig.2A).We then determinedwhetherthisMCR-mediatedinhibitionofcellviability wasassociatedwiththeinductionofapoptosis.TheresultsofDAPI stainingshowedthat MCRtreatmentsignificantlyincreasedthe numberofcondensedorblebbingnuclei,whichgenerallyappearin apoptosisbeforenuclearfragmentation(Fig.2B).Consistently,DNA fragmentationwasobservedbyMCRtreatmentat100g/mland
graduallyincreasedinaconcentration-dependentmanner(Fig.2C). MCRalsoenhancedthepopulationofannexinV+/PI–cells,which representsearlyapoptoticcells(Fig.2D).Inaddition,weexamined thequestionofwhetherornotMCRdisplayedtheantiproliferative effectsinhumannormallungfibroblastWI-38cells.Comparedwith untreatedcells,decreasedcellproliferationwasnotobservedin normallungWI-38cellstreatedwithMCR(Fig.1B).Thesedata col-lectivelysuggestthatMCRsuppressedcellproliferationbyinducing apoptosisinAGScells.
Bothextrinsicandintrinsicapoptosispathwayswereactivatedby MCTinAGScells
Giventhattherearetwoclassicalpathwaysinapoptosis,the extrinsicand intrinsic pathways, we examined which pathway isinvolved in MCR-inducedapoptosis. Our resultsshowedthat theexpressionsofTNF-relatedapoptosis-inducingligand(TRAIL), DR4,DR5,andFasligand(FasL)wereconcentration-dependently increasedby MCRtreatmenteven thoughtheexpressionof Fas wasnotchanged,suggestingthatMCRmightregulatetheextrinsic pathway(Fig.3A).Additionalimmunoblottingdataindicatedthat anti-apoptoticBcl-2expressionwasmoderatelydownregulatedby MCRtreatment,butMCRmarkedlyincreasedtheexpressionof pro-apoptoticBax(Fig.3B).Accordingly,MCRtreatmentreducedthe MMPinaconcentration-dependentmanner(Fig.3C),whichwas consistentwiththereleaseofcytochromecfromthemitochondria intothecytosol(Fig.3D), indicatingthatmitochondrial damage mayalsocontributetoMCR-inducedAGScellapoptosis.
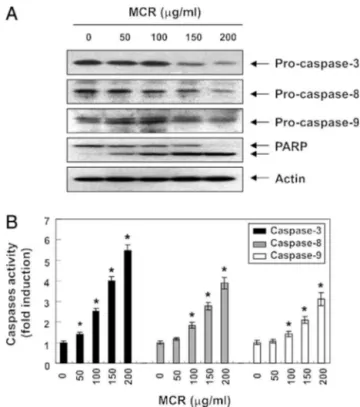
MCRinducedapoptosisbyactivatingcaspasesinAGScells
Theextrinsicandintrinsicsignalingpathwaysactivatecaspase cascades,whichisakeyhallmarkofapoptosis.Toexaminewhether MCRactivatescaspases,weinvestigatedtheexpressionand activ-ityoftwoinitiatorcaspasesoftheextrinsicandintrinsicapoptosis pathways,caspase-8and-9,respectively,andcaspase-3,atypical effectorcaspase.AsshowninFig.4A,MCRtreatmentapparently suppressedtheexpressionofpro-caspase-9,-8,and-3.The sub-sequentincreaseofcleavedpoly(ADP-ribose)polymerase(PARP) wasalsoobserved(Fig.4A).Consistently,theinvitroactivityofthe caspaseswassignificantlyenhancedbyMCRtreatment(Fig.4B). AlthoughwedidnotobservethetruncatedBid(tBid),whichmight
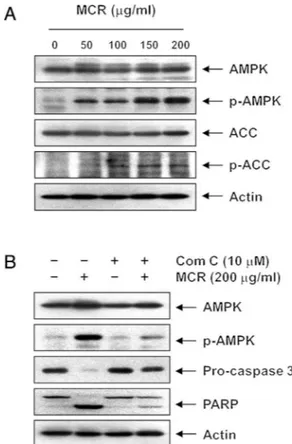
Fig.6.AMPKactivationbyMoutanCortexRadicistreatmentinAGScells.Thecells weretreatedwithMCRfor24h(A)orpre-treatedwithcompoundC(ComC)for1h andthentreatedwithMCRfor24h(B).Equalamountsofcelllysatewereresolved bySDS-polyacrylamidegels,transferredtoPVDFmembranes,andprobedwiththe indicatedantibodies.
beinvolvedintheregulationoftheextrinsicapoptosispathway (Lovell et al., 2008; Shamas-Din et al., 2011), MCR caused the progressivedownregulationofthetotalBidprotein(Fig.3B), pre-sumablyresultingfromtruncationbyactivatedcaspase-8.These resultsindicatethatthecytotoxiceffectsinducedbyMCRcould bemediatedthroughDR-mediatedapoptosis,therebyaccentuating crosstalkbetweentheintrinsicandextrinsicapoptosispathways. TheseresultsareinaccordancewiththosefromFig.3,suggesting thatMCRregulatedboththeextrinsicandintrinsicpathways.
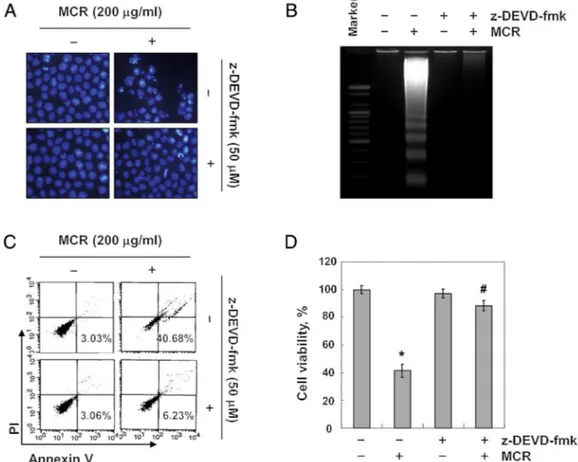
To verify whether MCR-induced apoptosis is caspase-dependent, we next inhibited the activity of caspase-3 using z-DEVD-fmk.OurresultsclearlyshowedthatMCR-induced apop-totic body formation and DNA fragmentation were absolutely abrogated by z-DEVD-fmk pre-treatment (Fig. 5A and B). The increasedpopulationofannexinV+/PI–cellsandthereducedcell viabilitywerealsoquitereversedbytheinhibitionofcaspase-3 activity(Fig.5CandD).Takentogether,ourresultsdemonstrate thattheactivationofcaspasecascadesisessentialforMCR-induced apoptosisinAGScells.
MCRinducedapoptosisbyactivatingAMPKinAGScells
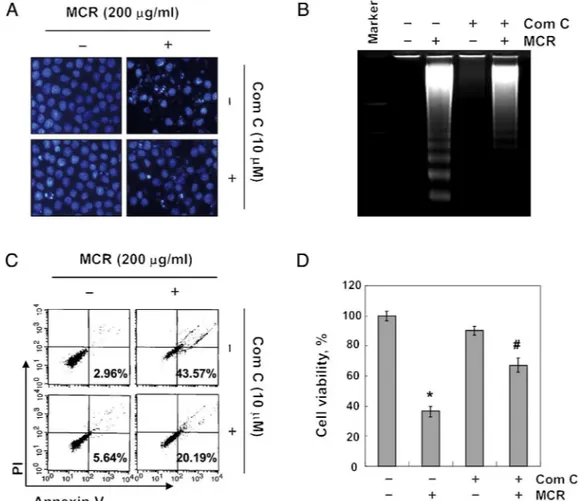
Fig.7.ThesuppressionofMoutanCortexRadicis-inducedapoptosisbytheinhibitionofAMPKinAGScells.Cellswerepre-treatedwithcompoundCfor1hbeforetreatment withMCRfor24h.(A)ThenucleiwerestainedwithDAPIsolutionandwerephotographed.(B)ThefragmentedDNAwasvisualizedunderUVlightafterstainingwithEtBr.(C) Thepercentagesofapoptoticcells(annexinV+/PI−cells)weremeasuredusingDNAflowcytometricanalysis.Dataarepresentedthemeanofthetwodifferentexperiments. (D)ThecellviabilitywasmeasuredbyanMTTassay.Thedataareexpressedasthemean±SDofthreeindependentexperiments(*p<0.05vs.untreatedcontrol;#p<0.05
vs.MCR-treatedcells).
thecleavageofcaspase-3anddegradationofPARPinMCR-treated cells,implyingalinkagebetweencaspaseandAMPKactivation.In linewiththeseobservations,themarkersofapoptosisincreasedby MCRtreatment,includingthecondensedandfragmentednuclei, DNAladder,andtheincreasedpopulationofannexinV+/PI−cells,
were significantly reversed by pretreatmentwith compound C (Fig.7A–C).Accordingly,theMCR-inducedreductionincell viabil-itywasrecoveredbytheadditionofcompoundC(Fig.7D).These observationscollectivelysuggest thatAMPKactivationplayed a crucialroleinMCR-inducedapoptosisinAGScellsandthatAMPK wasprobablyupstreamofcaspaseactivationinthesignaling path-wayinvolvedinthisprocess.
Discussion
Inthecurrent study,weinvestigatedtheanti-canceractivity ofMCRandexploredtheunderlyingmechanisminAGS human gastriccancercells.Althoughavarietyofresearchershavealready reportedontheanti-proliferativeeffectsofEvodiarutaecarpaand itscomponentsin variouscancercells,this isthefirststudy,to ourknowledge,toproposeAMPKasacriticalmoleculemediating MCR-inducedapoptosis.
Our resultsclearly showed that MCRinduced apoptotic cell deathinAGScells.MCRtreatmentmarkedlyincreasedtheratio of Bax/Bcl-2expression, a critical component of the mitochon-drialpathway,andsubsequentlytriggeredthelossofMMP,which wasassociatedwiththeincreasedexpressionofseveralDR-related
proteins.Themediatorsofmitochondria-mediatedapoptosishave been determinedby variouspreceding reports. The first candi-date released fromthe mitochondriaupon apoptotic stimuli is cytochrome c,an essential component of the respiratory chain (MacKenzieandClark,2008;Hensleyetal.,2013).Itforms apop-tosomewithapoptoticpeptidaseactivatingfactor-1(Apaf-1)and pro-caspase-9toactivatecaspase-9andtheclassicalcaspase cas-cade(CoryandAdams,2002;Brökeretal.,2005).Consistently,our resultsshowedthatMCRactivatedcaspase-9aswellas caspase-3 andenhanced thecytosolicrelease of cytochromec, together withthepronouncedactivationofcaspase-8andreductioninBid, suggesting that MCR-induced apoptosis occurredby accentuat-ing the crosstalk betweenthe intrinsic and extrinsic apoptosis pathways.Astheothercandidatesreleasedfromthe mitochon-dria to elicit apoptosis are apoptosis-inducing factor (AIF) and endonuclease G(endoG), which areparticularlyinvolvedin the caspase-independentpathway(Lietal.,2001;Creganetal.,2004). However,basedonourpresentdataindicatingthatz-DEVD-fmk completelyblockedtheapoptoticcelldeathinducedbyMCR,we suggestthatAIFandendoGmightonlyplayasmallrolein MCR-inducedapoptosis.
whichresultsinAMPKactivation(Panetal.,2004;Fiarolaetal., 2015).Inthecurrentstudy,MCRtreatmentofAGScellspromoted the concentration-dependent phosphorylation of AMPK and of ACC,itsdownstreamtarget.However,theMCR-induced phospho-rylationofbothAMPKandACCwasdiminishedmarkedlybyan inhibitorofAMPK,compoundC,suggestingthatMCRcouldbean AMPKactivator. Furthermore,the blockageof AMPK activation prevented the MCR-mediated induction of apoptosis and the reductionofcellviability,stronglysuggestingthatMCRactivated AMPK,whichledtocaspase-dependentcellapoptosisinAGScells. AlthoughAMPKmightgivecancercellsa survivaladvantage byconservingenergyinATP-depletedconditions(Laderouteetal., 2006;Rubinszteinetal.,2007),ithasbeengenerallyconsidered a tumorsuppressorbased onthe following points: first,AMPK deactivatesmTOR,whichiscommonlyactivatedinmanycancers; second,mostofthetumorsuppressorgeneshavebeenidentified asupstreamactivatorsofAMPK(Hardie,2004;Fengetal.,2007); third,AMPKactivatorshavebeenreportedtoinhibit tumorigene-sisaswellastumorgrowth(Buzzaietal.,2007;Saueretal.,2012; Suietal.,2014).Notably,metforminandantroquinonolhavebeen reportedtoactivateAMPKtoinduceapoptosisbyinducing mito-chondrialstress(Zakikhanietal.,2006;Chiangetal.,2010).AvicinD alsodisruptsmitochondrialmetabolism,leadingtodecreasedATP levelsandAMPKactivation,whichisfollowedbyautophagiccell death(Xuetal.,2007).Theseresultsareinaccordancewithour presentdataproposingAMPKasakeymoleculetoinduce apopto-sisinAGScellsandsupportthepossibilitythattheMCR-induced lossofMMPleadstoATPdepletionandthesubsequentactivation ofAMPK.However,furtherstudiesarewarrantedtodeterminethe preciseupstreamactivatorofAMPK.
Inconclusion,ourpresentresultsverifiedtheanti-cancereffects ofMCRinAGShumangastriccancercells.Wedemonstratedthat MCRinduces apoptosisin AGS cells via thecaspase-dependent extrinsicandintrinsicpathwaysbyincreasingDR-related regula-tors,MMPloss,andthereleaseofcytochromec,combinedwith anincrease in theBax/Bcl-2 ratio.We also suggest that AMPK alsoplayedapivotalroleinmitochondria-mediatedapoptosisin responsetoMCRtreatment.AlthoughtheactivecompoundofMCR andtheexactmechanismthroughwhichAMPKisactivatedshould befurtherelucidated,ourresultsproposeMCRasaprospective clinicaloptiontotreatothercancers.
Ethicaldisclosures
Protectionofhumanandanimalsubjects. Theauthorsdeclare
thatnoexperimentswereperformedonhumansoranimalsfor thisstudy.
Confidentialityofdata. Theauthorsdeclarethatnopatientdata
appearinthisarticle.
Righttoprivacyandinformedconsent. Theauthorsdeclarethat
nopatientdataappearinthisarticle.
Authors’contributions
CP, MHH, SHP and SHH contributed in running the labora-torywork,analysisof thedata anddraftedthepaper.GYK and SKMcontributedtocriticalreading ofthemanuscript. WJKand YHCdesignedthestudy,supervisedthelaboratoryworkand con-tributedtocriticalreadingofthemanuscript.Alltheauthorshave readthefinalmanuscriptandapprovedthesubmission.
Conflictsofinterest
Theauthorsdeclarenoconflictsofinterest.
Acknowledgements
ThisresearchwassupportedbyBasicScienceResearchProgram throughtheNationalResearch FoundationofKorea(NRF) grant funded by the Korea government(2015R1A2A2A01004633 and 2014R1A1A1008460)andtheFunctionalDistrictsoftheScience Beltsupportprogram,MinistryofScience,ICTandFuturePlanning,
RepublicofKorea.
References
Bröker,L.E.,Kruyt,F.A.,Giaccone,G.,2005.Celldeathindependentofcaspases:a review.Clin.CancerRes.11,3155–3162.
Buzzai,M.,Jones,R.G.,Amaravadi,R.K.,Lum,J.J.,DeBerardinis,R.J.,Zhao,F., Vio-llet,B.,Thompson,C.B.,2007.Systemictreatmentwiththeantidiabeticdrug metforminselectivelyimpairsp53-deficienttumorcellgrowth.CancerRes.67, 6745–6752.
Cai,J.,Chen,S.,Zhang,W.,Hu,S.,Lu,J.,Xing,J.,Dong,Y.,2014.Paeonolreverses paclitaxelresistanceinhumanbreastcancercellsbyregulatingtheexpression oftransgelin2.Phytomedicine21,984–991.
Chen,H.,Wang,J.P.,Santen,R.J.,Yue,W.,2015.Adenosinemonophosphateactivated proteinkinase(AMPK),amediatorofestradiol-inducedapoptosisinlong-term estrogendeprivedbreastcancercells.Apoptosis20,821–830.
Chiang,P.C.,Lin,S.C.,Pan,S.L.,Kuo,C.H.,Tsai,I.L.,Kuo,M.T.,Wen,W.C.,Chen,P.,Guh, J.H.,2010.Antroquinonoldisplaysanticancerpotentialagainsthuman hepato-cellularcarcinomacells:acrucialroleofAMPKandmTORpathways.Biochem. Pharmacol.79,162–171.
Choi,H.S.,Seo,H.S.,Kim,J.H.,Um,J.Y.,Shin,Y.C.,Ko,S.G.,2012.Ethanolextract ofPaeoniasuffruticosaAndrews(PSE)inducedAGShumangastriccancercell apoptosisviafas-dependentapoptosisandMDM2-p53pathways.J.Biomed.Sci. 19,82.
Cory,S.,Adams,J.M.,2002.TheBcl2family:regulatorsofthecellularlife-of-death switch.Nat.Rev.Cancer2,647–656.
Cregan,S.P.,Dawson,V.L.,Slack,R.S.,2004.RoleofAIFincaspase-dependentand caspase-independentcelldeath.Oncogene23,2785–2796.
Eom,D.W.,Lee,J.H.,Kim,Y.J.,Hwang,G.S.,Kim,S.N.,Kwak,J.H.,Cheon,G.J.,Kim,K.H., Jang,H.J.,Ham,J.,Kang,K.S.,Yamabe,N.,2015.Synergisticeffectofcurcuminon epigallocatechingallate-inducedanticanceractioninPC3prostatecancercells. BMBRep.48,461–466.
Fan,L.,Song,B.,Sun,G.,Ma,T.,Zhong,F.,Wei,W.,2013.Endoplasmicreticulum stress-inducedresistancetodoxorubicinisreversedbypaeonoltreatmentin humanhepatocellularcarcinomacells.PLoSONE8,e62627.
Feng,Z.,Hu,W.,deStanchina,E.,Teresky,A.K.,Jin,S.,Lowe,S.,Levine,A.J.,2007. TheregulationofAMPKbeta1,TSC2,andPTENexpressionbyp53:stress,cell andtissuespecificity,andtheroleofthesegeneproductsinmodulatingthe IGF-1-AKT-mTORpathways.CancerRes.67,3043–3053.
Figarola,J.L.,Singhal,J.,Tompkins,J.D.,Rogers,G.W.,Warden,C.,Horne,D.,Riggs, A.D.,Awasthi,S.,Singhal,S.S.,2015.SR4uncouplesmitochondrialoxidative phosphorylation,modulatesAMP-dependentkinase(AMPK)-mammaliantarget ofrapamycin(mTOR)signaling,andinhibitsproliferationofHepG2 hepatocar-cinomaCells.J.Biol.Chem.290,30321–30341.
Fu,P.K.,Wu,C.L.,Tsai,T.H.,Hsieh,C.L.,2012.Anti-inflammatoryand anticoagula-tiveeffectsofpaeonolonLPS-inducedacutelunginjuryinrats.Evid.Based Complement.Alternat.Med.2012,837513.
Fulda,S.,Debatin,K.M.,2006.Extrinsicversusintrinsicapoptosispathwaysin anti-cancerchemotherapy.Oncogene25,4798–4811.
Gao,Y.,He,C.,Ran,R.,Zhang,D.,Li,D.,Xiao,P.G.,Altman,E.,2015.Theresveratrol oligomers,cis-andtrans-gnetinH,fromPaeoniasuffruticosaseedsinhibitthe growthofseveralhumancancercelllines.J.Ethnopharmacol.169,24–33. Gwinn,D.M., Shackelford,D.B.,Egan,D.F.,Mihaylova,M.M.,Mery,A.,Vasquez,
D.S.,Turk,B.E.,Shaw,R.J.,2008.AMPKphosphorylationofraptormediatesa metaboliccheckpoint.Mol.Cell30,214–226.
Hardie, D.G., 2004. The AMP-activated protein kinase pathway-new players upstreamanddownstream.J.CellSci.117,5479–5487.
Hensley,P.,Mishra,M.,Kyprianou,N.,2013.Targetingcaspasesincancer therapeu-tics.Biol.Chem.394,831–843.
Hirai,A.,Terano,T.,Hamazaki,T.,Sajiki,J.,Saito,H.,Tahara,K.,Tamura,Y.,Kumagai, A.,1983.StudiesonthemechanismofantiaggregatoryeffectofMoutanCortex. Thromb.Res.31,29–40.
Jeon,S.M.,2016.RegulationandfunctionofAMPKinphysiologyanddiseases.Exp. Mol.Med.48,e245.
Kantari,C.,Walczak,H.,2011.Caspase-8andbid:caughtintheactbetweendeath receptorsandmitochondria.Biochim.Biophys.Acta1813,558–563. Kim,H.G.,Park,G.,Piao,Y.,Kang,M.S.,Pak,Y.K.,Hong,S.P.,Oh,M.S.,2014.Effects
Laderoute,K.R.,Amin,K.,Calaoagan,J.M.,Knapp,M.,Le,T.,Orduna,J.,Foretz,M., Viol-let,B.,2006.5′-AMP-activatedproteinkinase(AMPK)isinducedbylow-oxygen
andglucosedeprivationconditionsfoundinsolid-tumormicroenvironments. Mol.CellBiol.26,5336–5347.
Li,L.Y.,Luo,X.,Wang,X.,2001.EndonucleaseGisanapoptoticDNasewhenreleased frommitochondria.Nature412,95–99.
Li,M.,Tan,S.Y.,Wang,X.F.,2014.Paeonolexertsananticancereffectonhuman colo-rectalcancercellsthroughinhibitionofPGE2synthesisandCOX-2expression.
Oncol.Rep.32,2845–2853.
Lovell,J.F.,Billen,L.P.,Bindner,S.,Shamas-Din,A.,Fradin,C.,Leber,B.,Andrews, D.W.,2008.MembranebindingbytBidinitiatesanorderedseriesofevents culminatinginmembranepermeabilizationbyBax.Cell135,1074–1084. MacKenzie,S.H.,Clark,A.C.,2008.Targetingcelldeathintumorsbyactivating
cas-pases.Curr.CancerDrugTargets8,98–109.
Monteverde,T.,Muthalagu,N.,Port,J.,Murphy,D.J.,2015.Evidenceof cancer-promotingrolesforAMPKandrelatedkinases.FASEBJ.282,4658–4671. Mukudai,Y.,Kondo,S.,Shiogama,S.,Koyama,T.,Li,C.,Yazawa,K.,Shintani,S.,2013.
RootbarkextractsofJuncuseffususandPaeoniasuffruticosaprotectsalivarygland acinarcellsfromapoptoticcelldeathinducedbycis-platinum(II)diammine dichloride.Oncol.Rep.30,2665–2671.
Nieminen,A.I.,Eskelinen,V.M.,Haikala,H.M.,Tervonen,T.A.,Yan,Y.,Partanen, J.I.,Klefström,J.,2013.Myc-inducedAMPK-phosphop53pathwayactivates Baktosensitize mitochondrialapoptosis.Proc. Natl.Acad.Sci.U.S.A. 110, 1839–1848.
Pan, D.,Dong,J.,Zhang, Y.,Gao, X., 2004. Tuberoussclerosis complex:from Drosophilatohumandisease.TrendsCellBiol.14,78–85.
Park,J.,Kim,H.Y.,Lee,S.M.,2011.ProtectiveeffectsofMoutanCortexRadicisagainst acutehepatotoxicity.Afr.J.Tradit.Complement.Altern.Med.8,220–225. Peng,L.H.,Liu,S.,Xu,S.Y.,Chen,L.,Shan,Y.H.,Wei,W.,Liang,W.Q.,Gao,J.Q.,2013.
Inhibitoryeffectsofsalidrosideandpaeonolontyrosinaseactivityandmelanin synthesisinmouseB16F10melanomacellsandultravioletB-induced pigmen-tationinguineapigskin.Phytomedicine20,1082–1087.
Poon,T.Y.,Ong,K.L.,Cheung,B.M.,2011.Reviewoftheeffectsofthetraditional ChinesemedicineRehmanniaSixFormulaondiabetesmellitusandits compli-cations.J.Diabetes3,184–200.
Rho,S.,Chung,H.S.,Kang,M.,Lee,E.,Cho,C.,Kim,H.,Park,S.,Kim,H.Y.,Hong,M., Shin,M.,Bae,H.,2005.Inhibitionofproductionofreactiveoxygenspeciesand geneexpressionprofilebytreatmentofethanolextractofMoutanCortexRadicis inoxidativestressedPC12cells.Biol.Pharm.Bull.28,661–666.
Rubinsztein,D.C.,Gestwicki,J.E.,Murphy,L.O.,Klionsky,D.J.,2007.Potential thera-peuticapplicationsofautophagy.Nat.Rev.DrugDiscov.6,304–312. Sauer,H.,Engel,S.,Milosevic,N.,Sharifpanah,F.,Wartenberg,M.,2012.Activationof
AMP-kinasebyAICARinducesapoptosisofDU-145prostatecancercellsthrough generationofreactiveoxygenspeciesandactivationofc-junN-terminalkinase. Int.J.Oncol.40,501–508.
Shackelford,D.B.,Shaw, R.J.,2009.TheLKB1-AMPKpathway: metabolismand growthcontrolintumoursuppression.Nat.Rev.Cancer9,563–575. Shamas-Din,A.,Brahmbhatt,H.,Leber,B.,Andrews,D.W.,2011.BH3-onlyproteins:
orchestratorsofapoptosis.Biochim.Biophys.Acta1813,508–520.
Shaw,R.J.,Bardeesy,N.,Manning,B.D.,Lopez,L.,Kosmatka,M.,DePinho,R.A., Cant-ley,L.C.,2004.TheLKB1tumorsuppressornegativelyregulatesmTORsignaling. CancerCell6,91–99.
Sui,X.,Xu,Y.,Yang,J.,Fang,Y.,Lou,H.,Han,W.,Zhang,M.,Chen,W.,Wang,K.,Li, D.,Jin,W.,Lou,F.,Zheng,Y.,Hu,H.,Gong,L.,Zhou,X.,Pan,Q.,Pan,H.,Wang,X., He,C.,2014.Useofmetforminaloneisnotassociatedwithsurvivaloutcomes ofcolorectalcancercellbutAMPKactivatorAICARsensitizesanticancereffect of5-fluorouracilthroughAMPKactivation.PLOSONE9,e97781.
Tran,Q.,Lee,H.,Park,J.,Kim,S.H.,Park,J.,2016.Targetingcancermetabolism– revisitingtheWarburgeffects.Toxicol.Res.32,177–193.
Wang,S.C.,Tang,S.W.,Lam,S.H.,Wang,C.C.,Liu,Y.H.,Lin,H.Y.,Lee,S.S.,Lin,J.Y.,2012. AqueousextractofPaeoniasuffruticosainhibitsmigrationandmetastasisofrenal cellcarcinomacellsviasuppressingVEGFR-3pathway.Evid.BasedComplement. Alternat.Med.2012,409823.
Woods,A.,Johnstone,S.R.,Dickerson,K.,Leiper,F.C.,Fryer,L.G.,Neumann,D., Schlat-tner,U.,Wallimann,T.,Carlson,M.,Carling,D.,2003.LKB1istheupstreamkinase intheAMP-activatedproteinkinasecascade.Curr.Biol.13,2004–2008. Xu,Z.X.,Liang,J.,Haridas,V.,Gaikwad,A.,Connolly,F.P.,Mills,G.B.,Gutterman,
J.U.,2007.Aplanttriterpenoid,avicinD,inducesautophagybyactivationof AMP-activatedproteinkinase.CellDeathDiffer.14,1948–1957.
You,M.K.,Kim,M.S.,Jeong,K.S.,Kim,E.,Kim,Y.J.,Kim,H.A.,2016.Loquat(Eriobotrya japonica)leafextractinhibitsthegrowthofMDA-MB-231tumorsinnudemouse xenograftsandinvasionofMDA-MB-231cells.Nutr.Res.Pract.10,139–147. Yun,C.S.,Choi,Y.G.,Jeong,M.Y.,Lee,J.H.,Lim,S.,2013.MoutanCortexRadicisinhibits
inflammatorychangesofgeneexpressioninlipopolysaccharide-stimulated gin-givalfibroblasts.J.Nat.Med.67,576–589.