J. Braz. Chem. Soc. vol.21 número2
Texto
Imagem

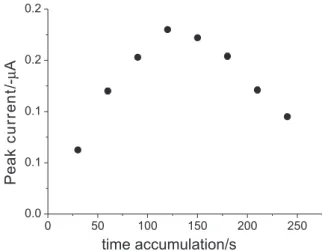
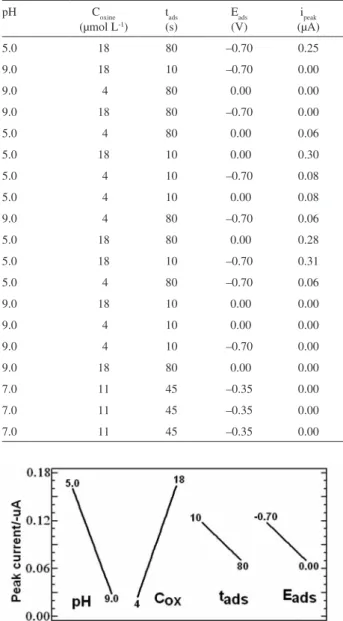
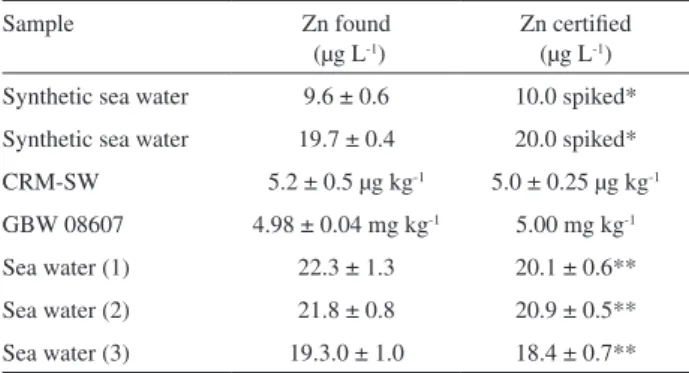
Documentos relacionados
study which was carried out in order to evaluate the efectiveness of maneuvers for swallowing in the activation of hyolaryngeal complex, through analysis of functional
The average of genotypic values of each progeny selected after selection of the best plants, general advances obtained through selection process carried out in this study,
(Apocynaceae) seeds from two ecosystems of northeastern Brazil (Caatinga and Restinga) and subjected to water stress in light and darkness at 30°C after 35 days.. Greek, capital
The probability of attending school four our group of interest in this region increased by 6.5 percentage points after the expansion of the Bolsa Família program in 2007 and
didático e resolva as listas de exercícios (disponíveis no Classroom) referentes às obras de Carlos Drummond de Andrade, João Guimarães Rosa, Machado de Assis,
Data on recapture of m.arked beetles (Cyclocephala hardyi).. Old fertilized flower of V. am azonica already becoming submerged for the fruit to develop
No phytosociological survey of vineyard weeds was carried out in Brazil and there was no detection of P. chlamydospora in vineyard weeds. Thus, the aims of the current study
Saída e city tour de Lugano uma das cidades mais encantadoras da suíça italiana, emoldurada pelo belo lago Lugano durante o nosso tour passaremos pela Piazza della
