Mining quorum sensing in pathogenic P. aeruginosa and C. albicans
Texto
Imagem

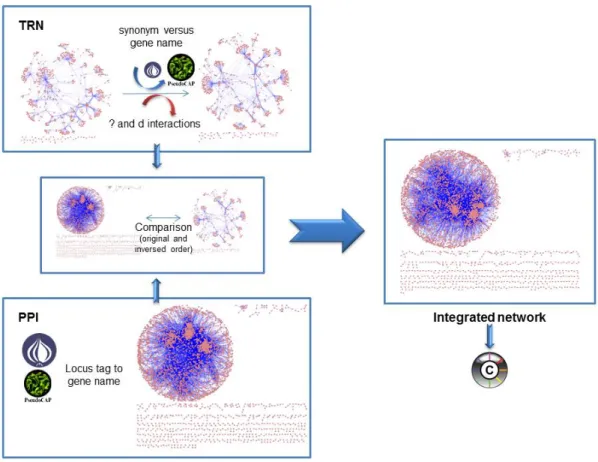
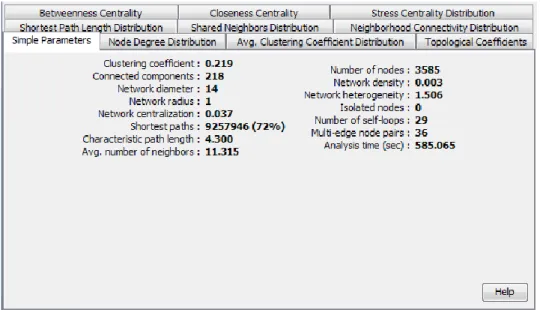

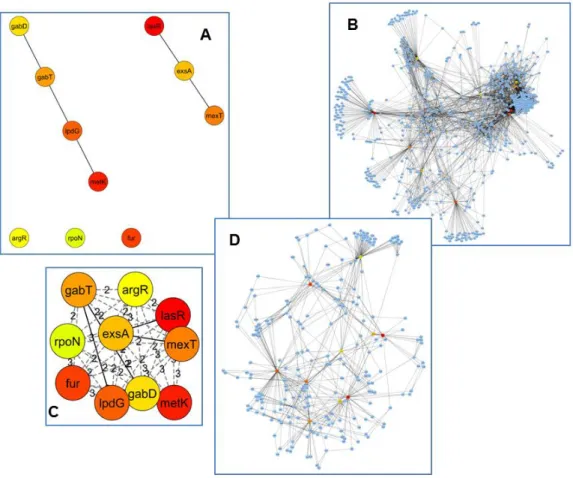



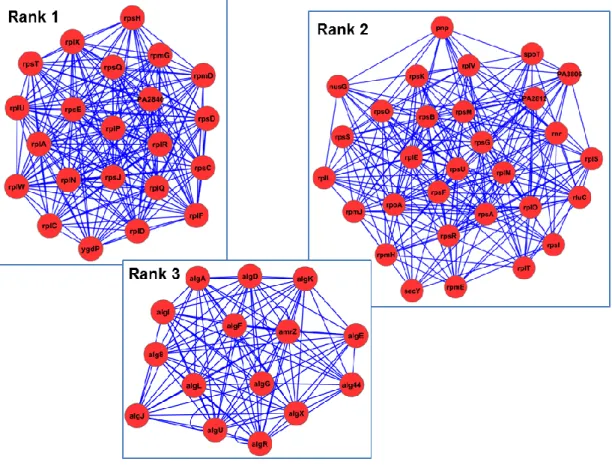
Documentos relacionados
Chronic hypoxia enhances endothelin-1-induced intracellular calcium ele- vation in rat carotid body chemoreceptors and up-regulates ETA receptor
Involvement of hippocampal AMPA glutamate receptor changes and the cAMP/protein kinase A/CREB-P signal- ling pathway in memory consolidation of an avoidance task in rats.. 1
This happens because the algorithm takes a period of time until all nodes have received the ideal jitter in the ACK frame, leading to a lower node throughput until that happens. When
The ANKK1 encodes a protein closely involved in the signal transduction pathway specially the DRD2, the gene responsible for the D2 dopamine subtype receptor.. The Taq1A
NIK has been described as a transmembrane signaling receptor that mediates an antiviral defense response based on the in vitro biochemical properties of the kinase, its inhibition
The canonical signaling protein in autophagy regulation is the mammalian target of rapamycin (mTOR), a ubiquitous protein kinase that is also involved in the regulation of cell
These pathways included T cell receptor signaling (hsa04660), Cytokine-cytokine receptor interac- tion (hsa04060), Measles (hsa05162), Jak-STAT signaling (hsa04630), Cell
Ao se analisar o gráfico da figura 3.9 nota-se uma clara tendência de queda do módulo de Weibull nos dois grupos de ensaio, ao inserir fibra de vidro na composição do material
