Cell Surface GRP78 Accelerated Breast
Cancer Cell Proliferation and Migration by
Activating STAT3
Xiaoli Yao1, Hua Liu2,3, Xinghua Zhang4, Liang Zhang1, Xiang Li1, Changhua Wang3, Shengrong Sun1*
1Department of Breast and Thyroid Surgery, Renmin Hospital of Wuhan University, Wuhan, 430060, Hubei Province, China,2Department of Clinical Pathology, The First People’s Hospital of Lianyungang,
Lianyungang, 222000, Jiangsu Province, China,3Department of Pathophysiology, Wuhan University School of Basic Medical Sciences, Wuhan, 430071, Hubei Province, China,4Department of Urology, Renmin Hospital of Wuhan University, Wuhan, 430060, Hubei Province, China
Abstract
High levels of cell surface glucose regulated protein 78 (sGRP78) have been implicated in cancer growth, survival, metastasis, and chemotherapy resistance. However, the underly-ing mechanism remains largely unknown. Here we report that the level of sGRP78 expres-sion in human breast tumors gradually increases during cancer progresexpres-sion.
Overexpression of GRP78 significantly enhanced its membrane distribution in human MCF-7 breast cancer cells, but had no effect on endoplasmic reticulum (ER) stress. High levels of sGRP78 facilitated cell proliferation and migration, as well as suppressed cell apoptosis. Neutralization of sGRP78 by a specific antibody against GRP78 alleviated sGRP78-in-duced cell growth and migration. Importantly, high phosphorylation levels of the signal transducer and activator of transcription 3 (STAT3) were found in human breast tumors that express sGRP78 and MCF-7 cells infected with adenovirus encoding human GRP78. Pre-treatment with a GRP78 antibody suppressed STAT3 phosphorylation. Furthermore, genet-ic and pharmacologgenet-ical inhibition of STAT3 reversed the impacts of GRP78 on cell
proliferation, apoptosis, and migration. These findings indicate that STAT3 mediates sGRP78-promoted breast cancer cell growth and migration.
Introduction
Glucose regulated protein 78 (GRP78, also known as binding immunoglobulin protein (BiP)) is a multi-functional protein predominantly expressed in the lumen of the endoplasmic reticu-lum (ER). Typically, GRP78 acts as a major ER chaperone and a master regulator of ER stress signaling through controlling protein folding and assembly, preventing protein aggregation, and regulating signaling of the unfolded protein response (UPR) [1–4]. As a central stress sen-sor, the level of GRP78 can be up-regulated by a variety of alterations in the tumor microenvi-ronment, such as hypoxia, glucose or nutrient deprivation, lactic acidosis, and inflammatory
OPEN ACCESS
Citation:Yao X, Liu H, Zhang X, Zhang L, Li X, Wang C, et al. (2015) Cell Surface GRP78 Accelerated Breast Cancer Cell Proliferation and Migration by Activating STAT3. PLoS ONE 10(5): e0125634. doi:10.1371/journal.pone.0125634
Academic Editor:Salvatore V Pizzo, Duke University Medical Center, UNITED STATES
Received:November 11, 2014
Accepted:March 16, 2015
Published:May 14, 2015
Copyright:© 2015 Yao et al. This is an open access article distributed under the terms of theCreative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Data Availability Statement:All relevant data are within the paper and its Supporting Information files.
response [5]. High levels of GRP78 promote cancer cell proliferation, survival, apoptosis resis-tance, immune escape, metastasis, angiogenesis in the microenvironment, and resistance to therapies [6,7]. Thus, GRP78 expression may serve as a biomarker for tumor behavior and treatment response, as well as a potential target for new therapies [6].
Currently, GRP78 was found to translocate to the surface of many types of cancer cells act-ing as an important regulator of oncogenic signalact-ing, cancer survival, and metastasis [5,8–10]. Particularly, the up-regulation of cell surface GRP78 (sGRP78), both at the RNA and protein level, presents in the cell membrane of malignant cells, but not in those of benign cells [8,11]. High levels of sGRP78 promote cancer cell proliferation, migration, apoptosis resistance, and invasion [12–14]. In contrast, neutralization of sGRP78 by a specific antibody against GRP78 suppresses tumor growth and metastasis bothin vitroandin vivo[10,15,16].
Signal transducer and activator of transcription 3 (STAT3) plays a vital role in cell survival and tumorigenesis [17,18]. STAT3 has been found to be constitutively activated in many cancers. Suppression of STAT3 by pharmacological agents and genetic interference inhibits cell proliferation, induces apoptosis, and suppresses tumorigenicityin vivo[17,18]. Thus, STAT3 may also be considered as a prognostic marker and therapeutic target in human breast cancer [19].
In the present study, we found that sGRP78 was highly expressed in breast tumors, accom-panied by the elevated STAT3 phosphorylation. Overexpression of GRP78 increased mem-brane distribution of GRP78 and enhanced STAT3 phosphorylation. Inhibition of sGRP78 function by a specific anti-GRP78 antibody mitigated GRP78-induced STAT3 phosphoryla-tion. Genetic and pharmacological inhibition of STAT3 abolished sGRP78-promoted breast cancer cell growth and migration. Our results, for the first time, suggest that sGRP78-induced tumor promotion is mediated by STAT3.
Materials and Methods
Material and reagents
Antibodies used in this study include the following: GRP78 antibody (N-20 and C-20, Santa Cruz Biotechnology, Santa Cruz, CA, USA), Phosphos-IRE1α(Ser724) antibody (Abcam, Cambridge, UK); E-cadherin (Stressgen, Victoria, Canada); STAT3, Phospho-STAT3 (Tyr705), JAK2, Phospho-JAK2 (Tyr1007/1008), CHOP, Caspase-3, IRE1α, and PARP anti-body (Cell Signaling Technology, Beverley, MD, USA); andβ-tubulin antibody (Sigma-Al-drich, Steinheim, Germany). Tunicamycin was from Sigma-Aldrich. Dulbecco’s Modified Eagles Medium (DMEM), fetal bovine serum (FBS), and Geneticin (G418) were purchased from HyClone (Logan, UT, USA). STAT3 specific inhibitor benzoic acid (2-Hydroxy-4-(((4-methylphenyl)sulfonyloxy)acetyl)amino)-benzoic acid, NSC74859), human STAT3/ shRNA, and control shRNA lentiviral particles were obtained from Santa Cruz Biotechnology.
Clinical Specimen and cell culture
The frozen breast tumor tissues and their paired adjacent non-tumor tissues were obtained from the Department of Clinic Pathology of Wuhan University Renmin Hospital. Written in-formed consent from the patients was obtained, and this series of studies was reviewed and ap-proved by Institutional Ethics Committees of Wuhan University Renmin Hospital.
Human MCF-7 and MDA-MB-453 breast cancer cells (ATCC, Manassas, VA, USA) were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C in a humidified atmosphere containing 5% CO2.
Cell proliferation assay
Cell proliferation was assessed in 96-well dishes using an 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described previously [20]. Briefly, 3×103 cells/well were seeded in 100μl of complete DMEM with 20% FBS. After treatment with or
without the indicated compounds for certain time, the medium was removed from each well, and then 150μl of fresh medium (without phenol red) with 50μl of 0.5 mg/ml MTT solution
was added. After incubation for 3 h at 37°C, the medium was carefully removed and 150μl of
MTT solvent was added into well. The plate was covered with tinfoil and agitated on an orbital shaker for 15 min. An ELISA plate reader (Biotek, Winooski, VT, USA) was used to determine absorbance at 590 nm with a reference filter of 620 nm.
Apoptosis assay
The cells were incubated in serum-free medium with or without 25 ng/ml of tumor necrosis fac-tor-alpha (TNFα) for 24 h, fixed with 3.8% paraformaldehyde for 20 min, and then stained with the terminal deoxyribonucleotidyl transferase (TdT)-mediated dUTP-digoxigenin nick end la-beling (TUNEL) reagent (Roche, Basel, Switzerland) for in situ apoptosis detection. Positive and negative controls were pretreated with 10 U/ml DNase or incubated without TdT, respectively. Apoptotic cells were detected under an Olympus FluoView FV1000 Confocal Microscope.
Wound migration assay
The cells were seeded into 6-well plates and cultured to 100% confluence. A pipette tip was used to make a straight scratch in the cell layer to create the wound. Then, the cells were washed with PBS, and treated with or without the indicated compounds. Mitomycin C (Sigma-Aldrich) was used to inhibit cell proliferation. Wound images were taken with a digital camera mounted on light microscope. The wound gap widths were measured using Image J software.
Adenovirus construction and virus infection
The construction of recombinant adenoviral vectors expressing human GRP78 (Ad/GRP78) were performed as previously described [21,22]. A recombinant adenoviral plasmid expressing
β-galactosidase (Ad/β-gal) was kindly provided by Dr. Xuejun Wang (The University of South Dakota Sanford School of Medicine, Vermillion, SD). Adenoviruses were amplified in HEK293 cells and purified with a cesium chloride gradient. MCF-7 and MDA-MB-453 cells were infected with Ad/GRP78 or a control Ad/β-gal (MOI:100,) in serum-free medium for 6 h, and then cul-tured in growth medium for 24 h. Lentivirus carrying human STAT3/shRNA was used to silence STAT3. Lentivirus transduction was performed according to the manufacturer's protocol.
Membrane fraction preparation and western blot
The cells and minced tissues were sonicated in ice cold lysis buffer (containing 50 mM Hepes, pH7.6, 150 mM NaCl, 1% Triton X-100, 1 mM NaF, 20 mM sodium pyrophosphate, 20 mMβ-glycerol phosphate, 1 mM sodium orthovanadate, 10μg/ml leupeptin, 10μg/ml
aprotinin, 1μM microcystin-LR, and 1mM phenylmethylsulfonyl fluoride) and put on ice for
proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a nitrocellulose membrane, and detected with specific antibod-ies.β-tubulin and E-cadherin served as the inner references of the cytosolic and membrane protein, respectively, for all western blots. The intensity of protein bands was quantified using Bio-Rad Quantity One software.
Immunofluorescence
The immunofluorescence labeling of formalin-fixed paraffin-embedded (FFPE) breast cancer tissues was performed as previously described [23]. Briefly, four-micron FFPE sections were heat-treated under pressure in a microwave for 5 min, and then incubated with primary anti-bodies for 1 h at room temperature. Next, the sections were incubated with secondary antibod-ies with fluorescent labels for 1 h at room temperature. The nuclei were stained with the DNA dye DAPI. The results were randomly evaluated independently by two specialized cancer-pa-thologists who were masked to all clinical data. The immunofluorescence results were analyzed with the Automated Quantitative Analysis (AQUA) system.
For staining of cultured cells, the cells were fixed with 3.7% paraformaldehyde, washed three times with PBS, and blocked with 1% bovine serum albumin (BSA, in PBS) for 1 h. After washing 3 times with PBS, the cells were incubated with primary antibody for 1 h at room tem-perature followed by washing 3 times with PBS and incubation with a second antibody conju-gated to fluorescent dyes for 1 h at room temperature. Finally, the mixture was mounted with mounting solution after three additional washes. Confocal images were taken by always using the same fixed scan parameters and fixed settings.
Statistical analysis
All values are represented as the mean ± SEM. Differences between mean values were exam-ined using the paired Student’st-test. Chi Square was used to test differences between two or more actual samples.p<0.05 was regarded as statistically significant. Linear regression was
cal-culated using Pearson’s correlation analysis. The quantification of the relative increase in pro-tein expression and phosphorylation statuses was normalized with control propro-tein expression in each experiment. The figures are representative of at least three independent experiments with similar results.
Results
Cell surface GRP78 is upregulated in human breast carcinoma
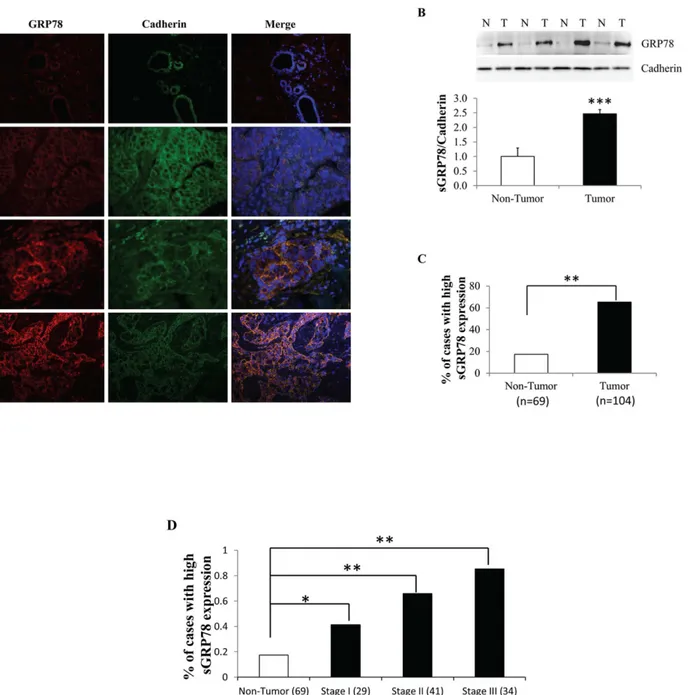
We firstly analyzed a series of paired human breast tumors and adjacent non-tumor tissues for the expression pattern of cell surface GRP78 (sGRP78) by immunofluorescence (IF). sGRP78 location was determined by co-localization with the membrane marker protein E-cadherin. The level of sGRP78 was assessed according to fluorescence intensity. Representative IF sec-tions showed that sGRP78 expression was gradually elevated with pathological tumor stage progression (Fig 1A). Western blot results (Fig 1B) also revealed that sGRP78 is highly express-ed in tumors when comparexpress-ed with a group of matchexpress-ed non-tumor counterparts, suggesting a positive correlation of sGRP78 with tumorigenesis. By scoring 69 non-tumor tissues and 104 tumor tissues, we found that sGRP78 was highly stained in 17.4% of non-tumor tissues and in 65.4% of tumor tissues (p<0.01) (Fig 1C). We next investigated the correlation of sGRP78 with
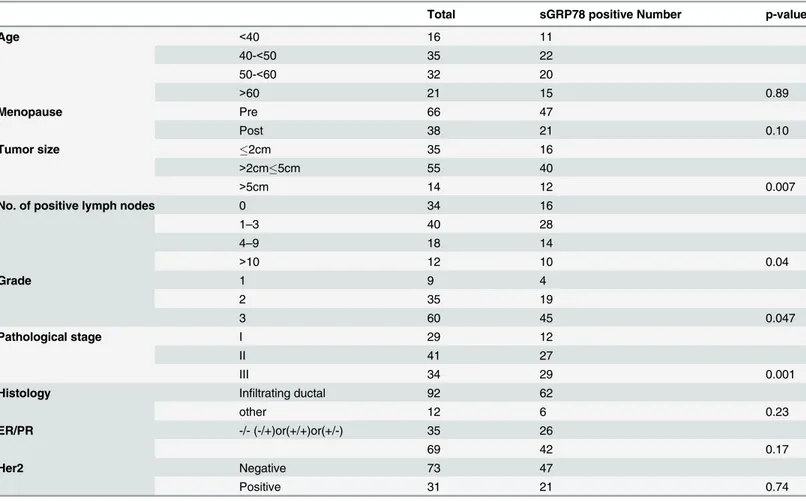
stage of breast cancer (Table 1). No relationship was observed between the sGRP78 expression and other characteristics such as age, menopause status, ER/PR expression, as well as Her2 ex-pression (Table 1). Thus, our results indicated that sGRP78 expression progressively increased during cancer progression.
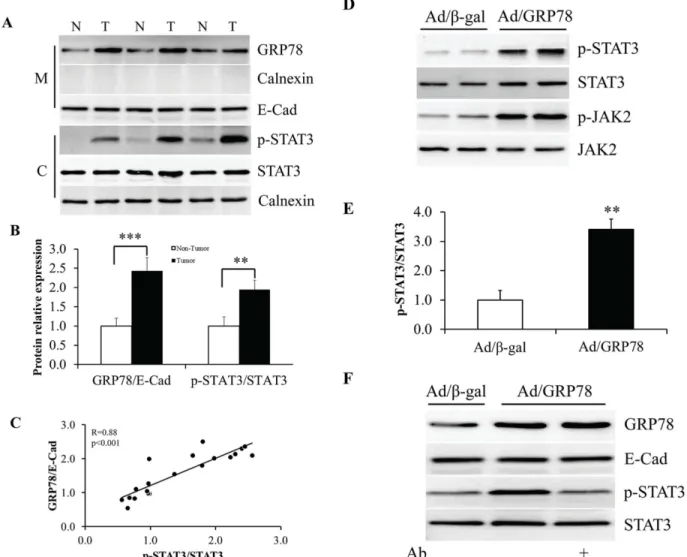
Fig 1. Association of sGRP78 expression with breast tumor progression.(A) Representative immunofluorescence staining of cell surface GRP78 (sGRP78) protein in sGRP78 positive human breast carcinomas. (B) Western blotting of sGRP78 protein in breast tumor and non-tumor tissues. The expression level of sGRP78 was normalized to E-cadherin and is represented in the bottom panel. (C) The percentage of sGRP78 highly stained cases in 104 tumor tissues and 69 non-tumor tissues detected by immunofluorescence analysis. (D) Distribution of sGRP78 highly stained cases with tumor characteristics.*p<0.05,**p<0.01,***p<0.001 vs the non-tumor group. N: non-tumor tissue; T: tumor tissue.
Cell surface GRP78 is up-regulated in MCF-7 cells infected with Ad/
GRP78
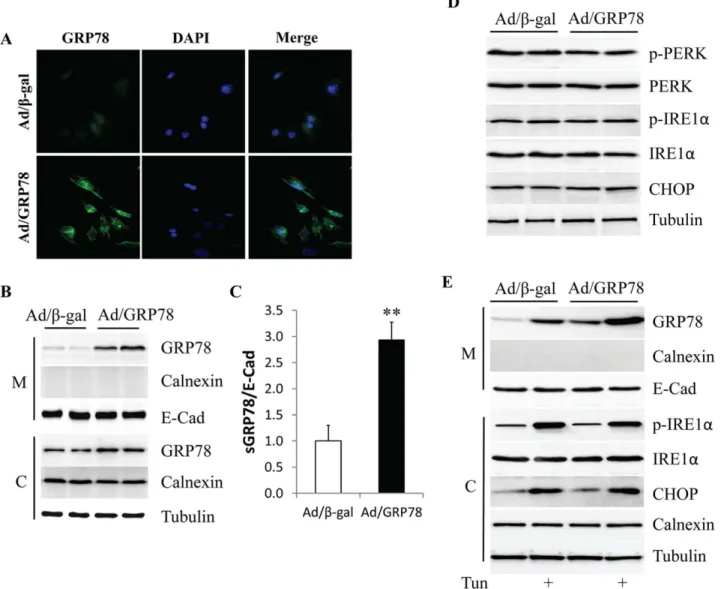
To observe the potential impact of GRP78 overexpression on its membrane distribution, GRP78 was overexpressed in MCF-7 cells which have the lowest endogenous levels of GRP78 protein [24], by infection with adenovirus encoding human GRP78 (Ad/GRP78). Adenovirus expressing
β-galactosidase (Ad/β-gal) served as the control. GRP78 expression was detected by immunoflu-orescence staining and western blotting, respectively. In non-permeabilized cells, staining pri-marily detects sGRP78, rather than intracellular GRP78. As shown inFig 2A, the
immunofluorescence intensity of sGRP78 was dramatically increased in the Ad/GRP78-infected MCF-7 cells when compared to the Ad/β-gal-infected cells. This result was further confirmed by western blot showing high GRP78 expression in the membrane fraction of the Ad/GRP78-in-fected MCF-7 cells (Fig 2B) or MDA-MB-453 cells (S1 Fig). When sGRP78 expression was nor-malized with the membrane marker protein E-cadherin, Ad/GRP78-infection drove a 2.9-fold increase in sGRP78 when compared with Ad/β-gal-infected MCF-7 cells (Fig 2C).
Considering that GRP78 is a master regulator involved in the regulation of ER stress and that ER stress is implicated in cancer survival, we next tested whether high sGRP78 expression is a consequence of ER stress resulting in GRP78 overexpression. As shown inFig 3D, GRP78 overexpression did not change the phosphorylation status of PERK and IRE1αand expression
Table 1. Association between sGRP78 expression and patient clinical/pathological characteristics.
Total sGRP78 positive Number p-value
Age <40 16 11
40-<50 35 22
50-<60 32 20
>60 21 15 0.89
Menopause Pre 66 47
Post 38 21 0.10
Tumor size 2cm 35 16
>2cm5cm 55 40
>5cm 14 12 0.007
No. of positive lymph nodes 0 34 16
1–3 40 28
4–9 18 14
>10 12 10 0.04
Grade 1 9 4
2 35 19
3 60 45 0.047
Pathological stage I 29 12
II 41 27
III 34 29 0.001
Histology Infiltrating ductal 92 62
other 12 6 0.23
ER/PR -/- (-/+)or(+/+)or(+/-) 35 26
69 42 0.17
Her2 Negative 73 47
Positive 31 21 0.74
P-values are based on chi-square test
of CHOP, indicating that ectopically expressed GRP78 did not induce ER stress. However, when MCF-7 cells were serum starved for 6 h, and then exposed to 2μg/ml of the ER stress
in-ducer tunicamycin for 24 h, sGRP78 expression was greatly increased in both Ad/β-gal- and Ad/GRP78-infected MCF-7 cells, suggesting that ER stress can upregulate the membrane dis-tribution of GRP78.
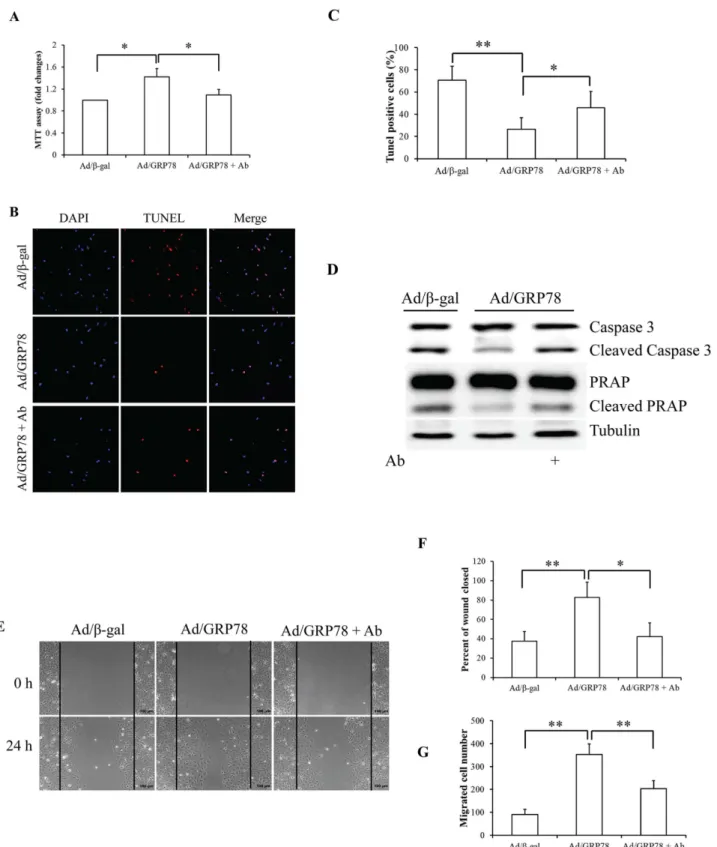
Cell surface GRP78 positively regulates cell growth and migration
To characterize the possible association of sGRP78 with cell proliferation and migration, we used the goat polyclonal antibody raised against a peptide mapping at the N-terminus of GRP78 of human origin (anti-GRP78 antibody,N-20, Santa Cruz) to block the sGRP78 recep-tor function [25–28]. Ad/β-gal- and Ad/GRP78-infected MCF-7 cells were incubated in growth medium with 4μg/ml anti-GRP78 antibody or normal goat IgG (Santa Cruz) for 24 h. The
Fig 2. Overexpression of GRP78 in MCF-7 cells.(A) Cell staining of sGRP78 in Ad/GRP78-infected MCF-7 cells. (B) Protein levels of sGRP78 as assessed by western blot. (C) Relative expression of sGRP78 in Ad/GRP78-infected MCF-7 cells is indicated as a fold change compared to control cells. (D) Effect of GRP78 overexpression on ER stress. (E) ER stress inducer tunicamycin stimulated sGRP78 expression.**p<0.01 vs Ad/β-gal-infected cells. M: membrane; C: cytosol; Tun: tunicamycin.
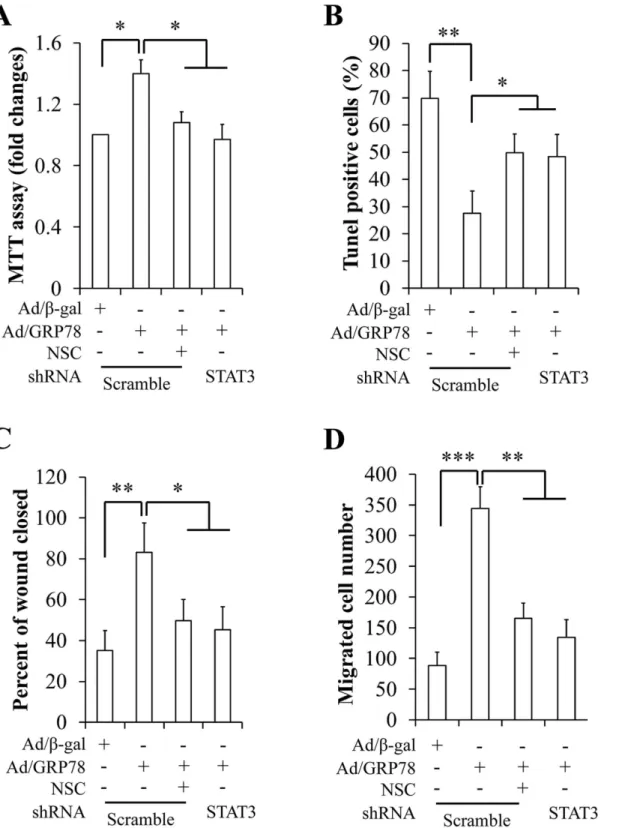
Fig 3. Effect of sGRP78 on cell proliferation, apoptosis, and migration.(A) Effect of sGRP78 on cell proliferation. (B) Cell apoptosis with TUNEL. (C) Quantitative analysis of cell apoptosis. (D) Western blotting of key apoptosis-associated protein expression. (E) Images of MCF-7 cells in the wound-healing migration assay. (F) Quantitative analysis of wound closure in areas as marked in (E). (G) Mean values for the number of migrated MCF-7 cells counted in the areas as marked in (E).*p<0.05,**p<0.01. Ab: Anti-GRP78 antibody.
isotype-specific normal goat IgG was used as control. An MTT assay was carried out to deter-mine cell proliferation. We found that GRP78 overexpression significantly increased cell prolif-eration and that GRP78-enhanced prolifprolif-eration was mitigated by treatment with GRP78 antibody (Fig 3A). To induce cell apoptosis, serum-starved cells were treated with 25 ng/ml of TNFαfor 24 h [29]. Cell apoptosis analysis by a TUNEL assay revealed that Ad/GRP78-in-fected cells were against TNFα-induced cell death, which is reversed by anti-GRP78 antibody
(Fig 3B). Expression of cleaved caspase 3 and PARP, two key apoptosis regulatory proteins,
supported that GRP78-suppressed apoptosis was deteriorated by anti-GRP78 antibody (Fig 3C). These data suggest that sGRP78 accelerates but neutralization of sGRP78 attenuates cell growth in MCF-7 cells.
Next, we used a wound-healing assay to determine the impact of sGRP78 on cell migration. Monolayer MCF-7 cells were treated with 4μg/ml of anti-GRP78 antibody or normal goat IgG
at 48 h after infection with Ad/β-gal and Ad/GRP78. Photographs were taken immediately after wound induction and following 24 h incubation. As shown in Fig3E–3G, we found that the wound healing abilities of GRP78-infected MCF-7 cells were markedly increased, com-pared with the control cells. In contrast, neutralization of sGRP78 by the anti-GRP78 antibody repressed wound healing capacity evidenced by decreased wound closure (Fig 3F) and number of migrated cells in the closed area (Fig 3G), suggesting that sGRP78 promotes cell migration.
Cell surface GRP78 stimulates JAK2/STAT3 pathway
To figure out the relationship between sGRP78 and STAT3, we next investigated STAT3 phos-phorylation in sGRP78 positive tumor tissues by western blot. We found that the tumor tissues with high expression of sGRP78 showed high levels of STAT3 phosphorylation (Fig 4A). Com-pared with adjacent non-tumor tissues, the cancer tissues show a ~2 fold increase of phosphor-ylated STAT3 (Fig 4B). A significant positive correlation was found with sGRP78 expression and STAT3 phosphorylation (r = 0.88, p<0.001;Fig 4C).
To address whether sGRP78 is responsible for increased STAT3 phosphorylation, MCF-7 cells were infected with Ad/β-gal and Ad/GRP78. The cells were harvested after 48 h of infec-tion and protein phosphorylainfec-tion was determined by western blot using specific antibody. We found that Ad/GRP78-infected cells showed enhanced phosphorylation of JAK2 and STAT3
(Fig 4D). Compared with Ad/β-gal infection, Ad/GRP78 infection resulted in ~3.4 fold increase
of the ratio of p-STAT3 to STAT3 (Fig 4E). When Ad/GRP78-infected cells were administrated with 4μg/ml of anti-GRP78 antibody for 24 h, the level of STAT3 phosphorylation dropped to
basal line (Fig 4FandS1 Fig). These results indicate that sGRP78 activates JAK2/STAT3 path-way in MCF-7 cells.
STAT3 mediates cell surface GRP78-induced action
We then investigated the impacts of STAT3 on sGRP78-induced cell proliferation, apoptosis, and migration. To suppress STAT3 phosphorylation, Ad/β-gal- and Ad/GRP78-infected MCF-7 cells were incubated with 100μM of a specific STAT3 inhibitor NSC74859 for 24 h or
and reduced the number of migrated cells (Fig 5D). Similarly, STAT3 knockdown significantly diminished effect of GRP78 overexpression on cell proliferation, apoptosis, and migration in MDA-MB-453 cells (S4 Fig). These data suggest that STAT3 might mediate sGRP78 function.
Discussion
Recent studies have established the critical roles of sGRP78 in the pathogenesis of cancer growth, metastasis, and resistance to chemotherapy [9,10,15,16]. In this study, we show that sGRP78 is a novel regulator in modulating cell growth and metastasis in breast cancer cells. Im-portantly, we confirm that sGRP78-induced activation of STAT3 is a key event in breast cancer cell proliferation and migration.
In agreement with early studies showing that GRP78 relocates to the cell membrane in ma-lignant but not in benign cells [8], we found that sGRP78 was highly expressed in human breast tumor, compared with adjacent non-tumor tissues (Fig1A–1C). In addition, our data
Fig 4. Stimulation of STAT3 phosphorylation by sGRP78.(A) Western blotting of sGRP78 protein and STAT3 phosphorylation in human breast tumor and non-tumor tissues. (B) Mean values of protein relative expression in (A). (C) Correlation of sGRP78 expression with STAT3 phosphorylation in human breast tumor. (D) STAT3 phosphorylation in Ad/GRP78-infected MCF-7 cells. (E) Quantitative analysis of STAT3 phosphorylation in (D). (F) Mitigation of sGRP78-induced STAT3 phosphorylation by treatment with anti-GRP78 antibody.*p<0.05,**p<0.01,***p<0.001. N: non-tumor tissue; T: tumor tissue; M: membrane; C: cytosol; Ab: Anti-GRP78 antibody.
Fig 5. Effect of genetic and pharmacologic inhibition of STAT3 on cell proliferation, apoptosis, and migration in MCF-7 cells.(A) Inhibition of STAT3 on cell proliferation. (B) Inhibition of STAT3 on cell apoptosis. (C) Inhibition of STAT3 on wound closure. (D) Inhibition of STAT3 on the number of migrated. *p<0.05,**p<0.01,***p<0.001, NSC: NSC74859, a specific STAT3 inhibitor.
demonstrates that the levels of sGRP78 were positively associated with pathological stage of breast cancer (Fig 1DandTable 1), suggesting that sGRP78 might contribute to breast cancer progression. This view is supported by ourin vitrostudies. Human MCF-7 breast cancer cells have the weakest invasive potential [30] and dormant characteristics [31], as well as the lowest endogenous levels of GRP78 protein expression in the cancer cell lines tested [24]. Our results found that overexpression of GRP78 in MCF-7 cells result in high-level expression and mem-brane distribution of GRP78 (Fig2A–2C), accompanied by significantly increased cell prolifer-ation, reduced cell apoptosis, and accelerated cell migration (Fig 3). This important impact of sGRP78 on the promotion of cancer cell growth and migration was further confirmed by using a specific antibody to inhibit sGRP78 function. Our results showed that immunoneutralization of sGRP78 attenuated the Ad/GRP78-induced impact on cell growth and migration (Fig 3), suggesting that sGRP78 is responsible for cancer cell growth and migration. Taken together, our findings strongly indicate that sGRP78 contributes to breast cancer survival and migration.
It has been reported that binding of N- and C-terminal anti-GRP78 antibodies (N-20 and C-20) generates distinct impacts on GRP78 signaling. The carboxyl terminal GRP78 antibodies (C-20) inhibit GRP78 signaling, while amino terminal GRP78 antibodies (N-20) stimulate GRP78 signaling [16,32]. In contrast, some studies have demonstrated that anti-GRP78 N-20 antibody can block sGRP78 functions effectively in many human cancers [25–28]. Our prelim-inary studies showed that either N-20 or C-20 antibody could impede sGRP78 signaling to-wards cell proliferation and migration (Data not shown). However, we do not understand the underlying mechanism. The further work is necessary to decipher why antibodies directed against these different GRP78 epitopes both impact STAT3 signaling.
It is well known that GRP78 plays a key role in the regulation and induction of ER stress [11,33], which has a profound effect on cancer cell proliferation and survival [33,34]. The question is whether sGRP78 stimulates cell growth and migration by ER stress. Our results found that the ER stress inductor tunicamycin dramatically increased the level of sGRP78 in MCF-7 cells (Fig 2E), consistent with previous observations that have demonstrated cell surface localization of GRP78 may be induced by thapsigargin in normal or cancer cells, including MCF-7 cells [35,36]. However, overexpression of GRP78 did not lead to ER stress or an un-folded protein response (UPR) (Fig 3D), suggesting that sGRP78 function is independent from ER stress.
STAT3 has been demonstrated to contribute to tumor growth through regulating protein expression of other signaling molecules, apoptosis, and the cell cycle [37]. In the present study, we found a positive correlation existed between sGRP78 levels and STAT3 phosphorylation in human breast cancer tissues (Fig4A–4C). The elevated phosphorylation of STAT3 and its up-stream kinases JAK2 was also expressed in Ad/GRP78-infected MCF-7 cells (Fig4Dand4E). Inhibition of sGRP78 functions by GRP78 antibody mitigated Ad/GRP78-induced STAT3 phosphorylation. These findings indicate that sGRP78 can activate the JAK2/STAT3 pathway. In addition, genetic and pharmacological inhibition of STAT3 significantly attenuated Ad/ GRP78-induced cell growth and migration (Fig 5andS4 Fig). Thus, our findings are intended to support a conclusion that STAT3 mediates sGRP78 action on breast cancer cell proliferation and migration.
sGRP78 functions as a receptor on the cell surface [38]. Through forming complexes with specific cell surface proteins, sGRP78 regulates signal transduction of cancer cells, such as the
antibody also did not cross-immunoprecipitate with GRP78 (data not shown), consistent with the previous study showing that STAT3 is not associated with GRP78 [43]. These results sug-gest that sGRP78 modulates the STAT3 pathway by an indirect manner.
The PI3K and STAT3 signaling pathways were historically regarded as two distinct regula-tory networks but emerging evidence suggests that the two pathways actually interact with each other and their interplay may be critical to promote tumor growth [44]. In some human tumor cell lines, the enhanced phosphorylation of STAT3 is inhibited by the PI3K inhibitor [45]. On the contrary, pretreatment with the JAK2/STAT3 pathway inhibitor AG490 or stattic inhibited propofol-induced AKT phosphorylation [5,46], suggesting that the link between PI3K and STAT3 pathways is significant in human cancers. Given that the dual activation and crosstalk of cytoprotective PI3K/AKT and JAK2/STAT3 pathways exists in sGRP78 positive cancer cells, it is possible that sGRP78-stimulated PI3K/AKT signaling is involved in the regu-lation of STAT3 phosphoryregu-lation. More studies are needed to further define this possibility.
In summary, our results demonstrate, for the first time, that sGRP78-dependent STAT3 ac-tivation increases breast cancer cell growth and migration. This finding might further lead to the development of new therapeutic approaches targeted against breast cancer with high level of sGRP78.
Supporting Information
S1 Fig. Overexpression of GRP78 in MDA-MB-453 cells.MDA-MB-453 cells were infected with Ad/GRP78 or Ad/β-gal in serum free medium for 4 h, and then incubated in growth me-dium. Forty-eight hours late after infection, the cells were cultured in the present or absent of 4μg/ml anti-GRP78 antibody or isotype-specific serum for 24 h. Membrane fraction was
iso-lated as described in Material and Methods. Western blot was performed to detect protein ex-pression and its phosphorylation by using specific antibodies. (A) Protein levels of sGRP78 as assessed by western blot. (B) Effect of GRP78 neutralization by anti-GRP787 antibody on JAK2/STAT3 pathway. Ab: anti-GRP78 antibody.
(TIF)
S2 Fig. NSC74859 suppressed GRP78-stimulated STAT3 phosphorylation in Ad/ GRP78-infected MCF-7 cells.MCF-7 cells were infected with Ad/GRP78 or Ad/β-gal in serum free medium for 4 h, and then incubated in growth medium. Forty-eight hours late after infection, MCF-7 cells were treated with 100μM of specific STAT3 inhibitor NSC74859 for 2 h
[47,48]. Western blot was performed to detect STAT3 protein expression and its phosphoryla-tion by using specific antibodies. (A) Representative western blot image of STAT3 phosphory-lation. (B) Quantification of phosphorylated STAT3 in (A). (C) Representative western blot image of irrelevant signaling molecules.p<0.05 vs Ad/β-gal control, ##p<0.01 vs Ad/
GRP78 group. (TIF)
S3 Fig. STAT3/shRNA lentiviral particles mitigated STAT3 expression and abolished STAT3 phosphorylation.MCF-7 cells were infected with human STAT3/shRNA and control shRNA lentiviral particles at 48 h after Ad/GRP78 or Ad/β-gal (as controls) infection. 48 hours later, the cells were harvested and western blot was performed to detect STAT3 protein expres-sion and its phosphorylation by using specific antibodies.
(TIF)
were assessed by MTT, TUNEL, and wound healing assay, respectively. (A) STAT3 knockdown on cell proliferation. (B) STAT3 knockdown on cell apoptosis. (C) STAT3 knockdown on wound closure. (D) STAT3 knockdown on the number of migrated MDA-MB-453 cells.
p<0.05 vs Ad/β-gal- and STAT3/Scrambled-infected cells; #p<0.05 vs
GRP78-overex-pressed and STAT3/Scrambled-infected cells. (TIF)
Acknowledgments
We greatly thank Dr. Xuejun Wang (The University of South Dakota Sanford School of Medi-cine) for his kind gift of Ad/β-gal plasmid. This study was partially supported by a NSFC grant to Dr. Changhua Wang (Grant NO: 81170790/H0718).
Author Contributions
Conceived and designed the experiments: SS. Performed the experiments: XY. Analyzed the data: XY CW. Contributed reagents/materials/analysis tools: XY HL XZ LZ XL. Wrote the paper: XY SS.
References
1. Banhegyi G, Baumeister P, Benedetti A, Dong D, Fu Y, Lee AS, et al. Endoplasmic reticulum stress. Annals of the New York Academy of Sciences. 2007; 1113:58–71. Epub 2007/05/08. doi:10.1196/ annals.1391.007PMID:17483206.
2. Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS letters. 2007; 581(19):3641–51. Epub 2007/05/08. doi:10.1016/j.febslet.2007.04.045PMID:17481612; PubMed Central PMCID: PMC2040386.
3. Little E, Ramakrishnan M, Roy B, Gazit G, Lee AS. The glucose-regulated proteins (GRP78 and GRP94): functions, gene regulation, and applications. Critical reviews in eukaryotic gene expression. 1994; 4(1):1–18. PMID:7987045.
4. Haas IG. BiP (GRP78), an essential hsp70 resident protein in the endoplasmic reticulum. Experientia. 1994; 50(11–12):1012–20. PMID:7988659.
5. Li Z. Glucose regulated protein 78: a critical link between tumor microenvironment and cancer hall-marks. Biochimica et biophysica acta. 2012; 1826(1):13–22. Epub 2012/03/20. doi:10.1016/j.bbcan. 2012.02.001PMID:22426159.
6. Lee AS. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer research. 2007; 67(8):3496–9. Epub 2007/04/19. doi:10.1158/0008-5472.CAN-07-0325PMID:17440054.
7. Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2013; 32(7):805–18. Epub 2012/04/18. doi:10. 1038/onc.2012.130PMID:22508478; PubMed Central PMCID: PMC3819728.
8. Roller C, Maddalo D. The Molecular Chaperone GRP78/BiP in the Development of Chemoresistance: Mechanism and Possible Treatment. Frontiers in pharmacology. 2013; 4:10. doi:10.3389/fphar.2013. 00010PMID:23403503; PubMed Central PMCID: PMC3568707.
9. Zhang Y, Tseng CC, Tsai YL, Fu X, Schiff R, Lee AS. Cancer cells resistant to therapy promote cell sur-face relocalization of GRP78 which complexes with PI3K and enhances PI(3,4,5)P3 production. PloS one. 2013; 8(11):e80071. Epub 2013/11/19. doi:10.1371/journal.pone.0080071PMID:24244613; PubMed Central PMCID: PMCPmc3823711.
10. Liu R, Li X, Gao W, Zhou Y, Wey S, Mitra SK, et al. Monoclonal antibody against cell surface GRP78 as a novel agent in suppressing PI3K/AKT signaling, tumor growth, and metastasis. Clinical cancer re-search: an official journal of the American Association for Cancer Research. 2013; 19(24):6802–11. Epub 2013/09/21. doi:10.1158/1078-0432.ccr-13-1106PMID:24048331.
11. Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. The Biochemical journal. 2011; 434(2):181–8. Epub 2011/02/12. doi:10. 1042/BJ20101569PMID:21309747; PubMed Central PMCID: PMC3353658.
13. Li Z, Zhang L, Zhao Y, Li H, Xiao H, Fu R, et al. Cell-surface GRP78 facilitates colorectal cancer cell mi-gration and invasion. The international journal of biochemistry & cell biology. 2013; 45(5):987–94. Epub 2013/03/15. doi:10.1016/j.biocel.2013.02.002PMID:23485528.
14. Misra UK, Payne S, Pizzo SV. Ligation of prostate cancer cell surface GRP78 activates a proprolifera-tive and antiapoptotic feedback loop: a role for secreted prostate-specific antigen. The Journal of bio-logical chemistry. 2011; 286(2):1248–59. Epub 2010/11/09. doi:10.1074/jbc.M110.129767PMID: 21056970; PubMed Central PMCID: PMC3020732.
15. Misra UK, Pizzo SV. Ligation of cell surface GRP78 with antibody directed against the COOH-terminal domain of GRP78 suppresses Ras/MAPK and PI 3-kinase/AKT signaling while promoting caspase acti-vation in human prostate cancer cells. Cancer biology & therapy. 2010; 9(2):142–52. Epub 2010/04/07. PMID:20368692.
16. Misra UK, Mowery Y, Kaczowka S, Pizzo SV. Ligation of cancer cell surface GRP78 with antibodies di-rected against its COOH-terminal domain up-regulates p53 activity and promotes apoptosis. Molecular cancer therapeutics. 2009; 8(5):1350–62. Epub 2009/05/07. doi:10.1158/1535-7163.MCT-08-0990 PMID:19417154.
17. Xiong A, Yang Z, Shen Y, Zhou J, Shen Q. Transcription Factor STAT3 as a Novel Molecular Target for Cancer Prevention. Cancers. 2014; 6(2):926–57. Epub 2014/04/20. doi:10.3390/cancers6020926 PMID:24743778; PubMed Central PMCID: PMCPmc4074810.
18. Siveen KS, Sikka S, Surana R, Dai X, Zhang J, Kumar AP, et al. Targeting the STAT3 signaling path-way in cancer: role of synthetic and natural inhibitors. Biochimica et biophysica acta. 2014; 1845 (2):136–54. Epub 2014/01/07. doi:10.1016/j.bbcan.2013.12.005PMID:24388873.
19. Clevenger CV. Roles and regulation of stat family transcription factors in human breast cancer. The American journal of pathology. 2004; 165(5):1449–60. Epub 2004/10/29. doi:10.1016/s0002-9440(10) 63403-7PMID:15509516; PubMed Central PMCID: PMCPmc1618660.
20. Zhu S, Yao F, Li WH, Wan JN, Zhang YM, Tang Z, et al. PKC?-dependent activation of the ubiquitin proteasome system is responsible for high glucose-induced human breast cancer MCF-7 cell prolifera-tion, migration and invasion. Asian Pacific journal of cancer prevention: APJCP. 2013; 14(10):5687–92. Epub 2013/12/03. PMID:24289563.
21. Satoh T, Furuta K, Tomokiyo K, Nakatsuka D, Tanikawa M, Nakanishi M, et al. Facilitatory roles of novel compounds designed from cyclopentenone prostaglandins on neurite outgrowth-promoting activ-ities of nerve growth factor. Journal of neurochemistry. 2000; 75(3):1092–102. Epub 2000/08/11. PMID:10936191.
22. Li PX, Wong J, Ayed A, Ngo D, Brade AM, Arrowsmith C, et al. Placental transforming growth factor-beta is a downstream mediator of the growth arrest and apoptotic response of tumor cells to DNA dam-age and p53 overexpression. The Journal of biological chemistry. 2000; 275(26):20127–35. Epub 2000/04/25. doi:10.1074/jbc.M909580199PMID:10777512.
23. Robertson D, Savage K, Reis-Filho JS, Isacke CM. Multiple immunofluorescence labelling of formalin-fixed paraffin-embedded (FFPE) tissue. BMC cell biology. 2008; 9:13. Epub 2008/03/28. doi:10.1186/ 1471-2121-9-13PMID:18366689; PubMed Central PMCID: PMCPmc2288605.
24. Yeung BH, Kwan BW, He QY, Lee AS, Liu J, Wong AS. Glucose-regulated protein 78 as a novel effec-tor of BRCA1 for inhibiting stress-induced apoptosis. Oncogene. 2008; 27(53):6782–9. Epub 2008/09/ 09. doi:10.1038/onc.2008.290PMID:18776923.
25. Davidson DJ, Haskell C, Majest S, Kherzai A, Egan DA, Walter KA, et al. Kringle 5 of human plasmino-gen induces apoptosis of endothelial and tumor cells through surface-expressed glucose-regulated protein 78. Cancer research. 2005; 65(11):4663–72. Epub 2005/06/03. doi: 10.1158/0008-5472.CAN-04-3426PMID:15930284.
26. Philippova M, Ivanov D, Joshi MB, Kyriakakis E, Rupp K, Afonyushkin T, et al. Identification of proteins associating with glycosylphosphatidylinositol- anchored T-cadherin on the surface of vascular endothe-lial cells: role for Grp78/BiP in T-cadherin-dependent cell survival. Molecular and cellular biology. 2008; 28(12):4004–17. Epub 2008/04/16. doi:10.1128/MCB.00157-08PMID:18411300; PubMed Central PMCID: PMC2423122.
27. Kelber JA, Panopoulos AD, Shani G, Booker EC, Belmonte JC, Vale WW, et al. Blockade of Cripto binding to cell surface GRP78 inhibits oncogenic Cripto signaling via MAPK/PI3K and Smad2/3 path-ways. Oncogene. 2009; 28(24):2324–36. Epub 2009/05/08. doi:10.1038/onc.2009.97PMID: 19421146; PubMed Central PMCID: PMC2749668.
28. Burikhanov R, Zhao Y, Goswami A, Qiu S, Schwarze SR, Rangnekar VM. The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell. 2009; 138(2):377–88. Epub 2009/07/28. doi:10. 1016/j.cell.2009.05.022PMID:19632185; PubMed Central PMCID: PMC2774252.
pharmacology. 2001; 133(4):467–76. doi:10.1038/sj.bjp.0704093PMID:11399663; PubMed Central PMCID: PMC1572806.
30. Mukhopadhyay R, Theriault RL, Price JE. Increased levels of alpha6 integrins are associated with the metastatic phenotype of human breast cancer cells. Clinical & experimental metastasis. 1999; 17 (4):325–32. PMID:10545019.
31. Barkan D, Kleinman H, Simmons JL, Asmussen H, Kamaraju AK, Hoenorhoff MJ, et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer research. 2008; 68(15):6241–50. doi:10.1158/0008-5472.CAN-07-6849PMID:18676848; PubMed Central PMCID: PMC2561279.
32. de Ridder GG, Ray R, Pizzo SV. A murine monoclonal antibody directed against the carboxyl-terminal domain of GRP78 suppresses melanoma growth in mice. Melanoma research. 2012; 22(3):225–35. Epub 2012/04/13. doi:10.1097/CMR.0b013e32835312fdPMID:22495669.
33. Clarke HJ, Chambers JE, Liniker E, Marciniak SJ. Endoplasmic reticulum stress in malignancy. Cancer cell. 2014; 25(5):563–73. doi:10.1016/j.ccr.2014.03.015PMID:24823636.
34. Scriven P, Coulson S, Haines R, Balasubramanian S, Cross S, Wyld L. Activation and clinical signifi-cance of the unfolded protein response in breast signifi-cancer. British journal of signifi-cancer. 2009; 101(10):1692– 8. Epub 2009/10/29. doi:10.1038/sj.bjc.6605365PMID:19861963; PubMed Central PMCID:
PMC2778547.
35. Zhang Y, Liu R, Ni M, Gill P, Lee AS. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. The Journal of biological chemistry. 2010; 285 (20):15065–75. doi:10.1074/jbc.M109.087445PMID:20208072; PubMed Central PMCID: PMC2865300.
36. Delpino A, Castelli M. The 78 kDa glucose-regulated protein (GRP78/BIP) is expressed on the cell membrane, is released into cell culture medium and is also present in human peripheral circulation. Bio-science reports. 2002; 22(3–4):407–20. Epub 2003/01/09. PMID:12516782.
37. Wang XH, Liu BR, Qu B, Xing H, Gao SL, Yin JM, et al. Silencing STAT3 may inhibit cell growth through regulating signaling pathway, telomerase, cell cycle, apoptosis and angiogenesis in hepatocellular car-cinoma: potential uses for gene therapy. Neoplasma. 2011; 58(2):158–71. Epub 2011/02/01. PMID: 21275467.
38. Gonzalez-Gronow M, Selim MA, Papalas J, Pizzo SV. GRP78: a multifunctional receptor on the cell surface. Antioxidants & redox signaling. 2009; 11(9):2299–306. Epub 2009/04/01. doi:10.1089/ARS. 2009.2568PMID:19331544.
39. Misra UK, Pizzo SV. Activated alpha2-macroglobulin binding to cell surface GRP78 induces T-loop phosphorylation of Akt1 by PDK1 in association with Raptor. PloS one. 2014; 9(2):e88373. Epub 2014/ 02/12. doi:10.1371/journal.pone.0088373PMID:24516643; PubMed Central PMCID:
PMCPmc3916429.
40. Gray PC, Vale W. Cripto/GRP78 modulation of the TGF-beta pathway in development and oncogene-sis. FEBS letters. 2012; 586(14):1836–45. Epub 2012/02/07. doi:10.1016/j.febslet.2012.01.051PMID: 22306319; PubMed Central PMCID: PMC3723343.
41. Shani G, Fischer WH, Justice NJ, Kelber JA, Vale W, Gray PC. GRP78 and Cripto form a complex at the cell surface and collaborate to inhibit transforming growth factor beta signaling and enhance cell growth. Molecular and cellular biology. 2008; 28(2):666–77. Epub 2007/11/10. doi:10.1128/MCB. 01716-07PMID:17991893; PubMed Central PMCID: PMC2223411.
42. Misra UK, Pizzo SV. Receptor-recognized alpha(2)-macroglobulin binds to cell surface-associated GRP78 and activates mTORC1 and mTORC2 signaling in prostate cancer cells. PloS one. 2012; 7 (12):e51735. Epub 2012/12/29. doi:10.1371/journal.pone.0051735PMID:23272152; PubMed Central PMCID: PMCPmc3522726.
43. Guo GG, Patel K, Kumar V, Shah M, Fried VA, Etlinger JD, et al. Association of the chaperone glucose-regulated protein 58 (GRP58/ER-60/ERp57) with Stat3 in cytosol and plasma membrane complexes. Journal of interferon & cytokine research: the official journal of the International Society for Interferon and Cytokine Research. 2002; 22(5):555–63. Epub 2002/06/13. doi:10.1089/10799900252982034 PMID:12060494.
44. Vogt PK, Hart JR. PI3K and STAT3: a new alliance. Cancer discovery. 2011; 1(6):481–6. Epub 2012/ 02/22. doi:10.1158/2159-8290.cd-11-0218PMID:22348200; PubMed Central PMCID:
PMCPmc3279943.
46. Shravah J, Wang B, Pavlovic M, Kumar U, Chen DD, Luo H, et al. Propofol mediates signal transducer and activator of transcription 3 activation and crosstalk with phosphoinositide 3-kinase/AKT. Jak-Stat. 2014; 3:e29554. Epub 2014/08/12. doi:10.4161/jkst.29554PMID:25105067; PubMed Central PMCID: PMC4124059.
47. Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104 (18):7391–6. Epub 2007/04/28. doi:10.1073/pnas.0609757104PMID:17463090; PubMed Central PMCID: PMC1863497.