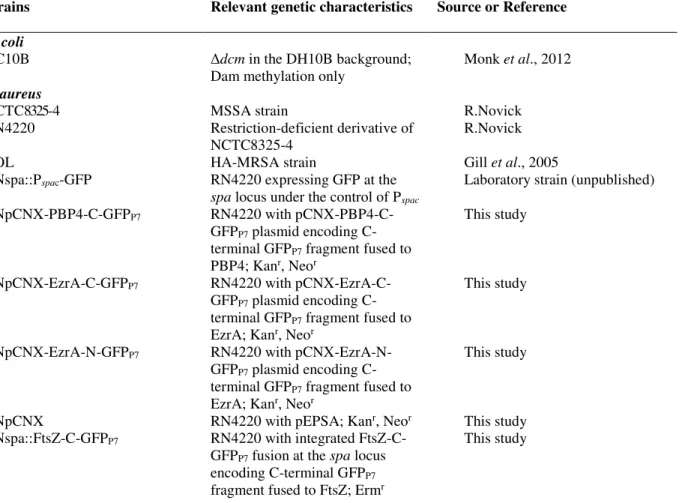
Study of in vivo interactions between penicillin-binding proteins of Staphylococcus aureus
Texto
Imagem





Documentos relacionados
Considering that the objectives of this work were to study the crystallization of proteins in the presence of AuNPs and their structural interactions with the AuNPs and gold atoms,
Desta forma, diante da importância do tema para a saúde pública, delineou-se como objetivos do es- tudo identificar, entre pessoas com hipertensão arterial, os
A total of 187 individual protein spots were detected, including spots of both resistant and susceptible strains, of which 58 proteins were found to be differentially expressed
In order to dissect the structural elements within the intrinsically disordered E7N domain, we recombinantly expressed or synthe- sized a series of proteins and peptide
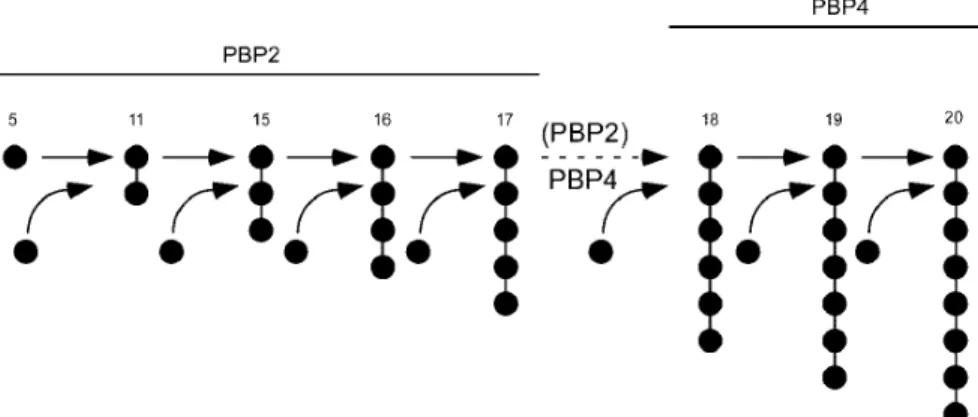
We suggest that protein interactions of PBP2 that occur via the N-terminal domain must be important for retaining PBP2 and/or other cell wall synthetic proteins in
Several of the 269 differen- tially expressed genes identified in this study encode for important proteins in oxidative pathways and liver metabo- lism and were expressed at
Inclui os seguintes domínios: domínio físico (dor, energia e fadiga, atividade sexual, sono, descanso e funções sensoriais ótimas); domínio psicológico (sentimentos
HTF-Rab27a proteins were purified from these cell lines by sequential immunoprecipitations (sequential steps depicted by Western blot in Figure 1 B) and co-purified proteins
