Vol-7, Special Issue3-April, 2016, pp1038-1045 http://www.bipublication.com
Research Article
In Silico
Determination and Validation of FptA Structure and Ligand Binding
Site as a vaccine candidate in
Pseudomonas aeruginosa
Sajjad Masoumi 1and Fateme Sefid2* 1Departeman of biology,
Science and Art University, Yazd, Iran Email:[email protected]
*2
Department of Biology, shahed University , Tehran-Qom Express Way
E-mail: [email protected]
* *Corresponding author, Biology Department,
Shahed University, Tehran-Qom Express way, Iran. Email: [email protected] [email protected]
ABSTRACT
Iron is an essential element for most living organisms. In mammalian hosts, iron is bound to proteins (hemoglobin, myoglobin, etc.) or stored within high-affinity iron molecules (transferrin, lactoferrin). Some of the bacterial pathogens release siderophore molecules in the external environment that scavenge iron from the proteins of the host and make it available for the bacteria. Iron–siderophore complexes are recognized by specific receptors embedded in the outer membrane. It has been shown that Pseudomonas aeruginosa(an opportunistic bacterium infecting a broad range of organisms) releases two major siderophores, which act also as virulence factors, i.e. pyochelin (Pch) and pyoverdine (Pvd). In comparsion with the Pvd and its outer membrane receptor FpvA, Pch transport and its receptor FptA (Mr: 75,993 Da) has been studied much little. Due to the important role of Pseudomonae in human infections, we have carried on the structural studies of FptA.
Key Words:pyochelin receptor, FptA, Pseudomonas aeruginosa
1. INTRODUCTION
Iron is an essential element for most living organisms[1]. In mammalian hosts, iron is bound to proteins (hemoglobin, myoglobin, etc.) or stored within high-affinity iron molecules (transferrin, lactoferrin). Some of the bacterial pathogens release siderophore molecules in the external environment that scavenge iron from the proteins of the host and make it available for the bacteria. Iron–siderophore complexes are recognized by specific receptors embedded in the outer membrane[2,3].It has been shown that Pseudomonas aeruginosa(an opportunistic bacterium infecting a broad range of organisms) releases two major siderophores, which act also as virulence factors, i.e. pyochelin (Pch) and pyoverdine (Pvd)[4,5]. In comparsion with the
implicated in both Co(II) and Mo(IV) delivery to P. aeruginosa cells[14]. Thus, this broad range of utilization makes this siderophore an attractive candidate for the design of new anti-microbial drugs that would use the Pch uptake pathway. The fpta gene coding for the Pch receptor FptA has been identified and characterized first in P. aeruginosa and more recently in B. cepacia[15]. The FptA amino acid sequence shares significant identity with a large number of TonB-dependent receptors (19.3% and 22.7% with FepA and BtuB from Escherichia coli and 32.5% with FpvA from P. aeruginosa), indicating a common mechanism of iron uptake with this family of transporters[16]. Due to the important role of Pseudomonae in human infections, we have carried on the structural studies of FptA.
2. METHODS
2.1. Sequence availability and homology search
The FptA protein sequence with. Accession AAC43213.1 GI 454353acquired from NCBI at http://www.ncbi.nlm.nih.gov/protein was saved in FASTA format for further analyses. The sequences served as a query for protein BLASTat http://blast.ncbi.nlm.nih.gov/Blast.cgi against non redundant protein database. Probable putative conserved domains of the query protein were also searched for, at the above address.
2.2. Primary sequence analysis
Protparam online software at http://expasy.org/tools/protparam.html was employed for estimation and determination of properties such as molecular weight, theoretical pI, amino acid composition, total number of negatively and positively charged residues, instability index and aliphatic index.
2.3. 3D structure prediction
The SWISS-MODEL Workspace at http://swissmodel.expasy.org/ is a web-based integrated service dedicated to protein structure homologymodelling. It assists and guides the user in building protein homology models at different levels of complexity. Building a homology model comprises four main steps: identification of
structural template(s), alignment of target sequence and template structure(s), model building, and model quality evaluation. These steps can be repeated until a satisfying modelling result is achieved. Each of the four steps requires specialized software and access to up-to-date protein sequence and structure databases.
2.4. Ligand binding site predictions
Cofactor
athttp://zhanglab.ccmb.med.umich.edu/COFACT OR/ is a structure-based method for biological function annotation of protein molecules. Important amino acid involved in ligand binding site is predicted by this server.
2.5. Pocket detection
DogSiteScorer at http://dogsite.zbh.uni-hamburg.de/ is an automated pocket detection and analysis tool which can be used for protein drugability assessment. Predictions with DoGSiteScorer are based on calculated size, shape and chemical features of automatically predicted pockets, incorporated into a support vector machine for druggability estimation.
2.6. Identification of functionally and structurally important residues
InterProSurf at http://curie.utmb.edu/pattest9.html predicting functional sites on protein surface using patch analysis was employed. FptA 3D structure, served as an input file for this server.
3. RESULT AND DISCUSSION
3.1. Sequence availability and homology search
The protein sequence with 720 residues obtained from NCBI and saved in FASTA format. Protein sequence serving as query for BLAST produced a set of sequences as the highest similar sequence. BLAST search revealed numerous hits to the FptA sequence. All hits were of Pseudomonas aeruginosa. Putative conserved domains were detected within this sequence and are shown in
Figure 1. TonB dependent/Ligand-Gated channels
barrel. Energy (proton-motive force) and TonB-dependent conformational alteration of channel (parts of plug, and loops 7 and 8) allow passage of ligand. FepA residues 12-18 form the TonB box, which mediates the interaction with the TonB-containing inner membrane complex. TonB preferentially interacts with ligand-bound receptors. Transport thru the channel may resemble passage thru an air lock. In this model, ligand binding leads to closure of the extracellular end of pore, then a TonB-mediated signal
facillitates opening of the interior side of pore, deforming the N-terminal plug and allowing passage of the ligand to the periplasm. Such a mechanism would prevent the free diffusion of small molecules thru the pore. TonB-dependent siderophorereceptor ;This subfamily model encompasses a wide variety of TonB-dependent outer membrane siderophore receptors. It has no overlap with TonB receptors known to transport other substances, but is likely incomplete due to lack of characterizations.
Figure 1.Putative conserved domains have been detected.
3.2. Primary sequence analysis
The protein sequence served as input for the computation of various physical and chemical parameters. The computed parameters included the molecular weight, theoretical pI, instability index, aliphatic index and grand average of hydropathicity (indicates the solubility of the proteins: positive GRAVY (hydrophobic), negative GRAVY (hydrophilic)) are summarized below.
Number of amino acids: 720
Molecular weight: 79992.0
Theoretical pI: 5.86
Aminoacid---composition:
Ala (A) 54 7.5% Arg (R) 51 7.1% Asn (N) 31 4.3% Asp (D) 46 6.4% Cys (C) 1 0.1% Gln (Q) 38 5.3% Glu (E) 34 4.7% Gly (G) 73 10.1% His (H) 9 1.2% Ile (I) 17 2.4%
Leu (L) 71 9.9% Lys (K) 22 3.1% Met (M) 9 1.2% Phe (F) 23 3.2% Pro (P) 42 5.8% Ser (S) 49 6.8% Thr (T) 53 7.4% Trp (W) 17 2.4% Tyr (Y) 38 5.3% Val (V) 42 5.8% Pyl (O) 0 0.0% Sec (U) 0 0.0%
Total number of negatively charged residues (Asp + Glu): 80
Total number of positively charged residues (Arg + Lys): 73
Atomic composition:
Carbon C 3559 Hydrogen H 5449 Nitrogen N 999 Oxygen O 1090 Sulfur S 10
Extinction coefficients:
Extinction coefficients are in units of M-1 cm-1, at 280 nm measured in water.
Ext. coefficient 150120
Abs 0.1% (=1 g/l) 1.877, assuming all pairs of Cys residues form cystines
Ext. coefficient 150120
Abs 0.1% (=1 g/l) 1.877, assuming all Cys residues are reduced
Estimated half-life:
The N-terminal of the sequence considered is M (Met).
The estimated half-life is: 30 hours (mammalian reticulocytes, in vitro).
>20 hours (yeast, in vivo).
>10 hours (Escherichia coli, in vivo).
Instability index:
The instability index (II) is computed to be 31.49
This classifies the protein as stable.
Aliphatic index: 72.08
Grand average of hydropathicity (GRAVY): -0.554
3.3. 3D structure prediction
Building a homology model comprises four main steps: identification of structural template(s), alignment of target sequence and template structure(s), model building, and model quality evaluation. These steps can be repeated until a satisfying modelling result is achieved. Each of the four steps requires specialized software and access to up-to-date protein sequence and structure databases. Swiss model software recruited for homology modeling introduced 1 model. Predicted model is shown in figureFigure 2.
Figure 2. 3D structure of FptA
3.4. Ligand binding site predictions
Ligand binding sites determined using COFACTOR software, indicate involvement of conserved residues include 59, 65 ,67, 72, 643, 644 in binding site with the highest CscoreLB (the confidence score of predicted binding site) (Figure 3).The calculated BS-score for this predicted binding site was 1.80.BS-score
Figure3.FptA ligand binding site predictions.
Ran k
CscoreL B
PDB Hit
TM-scor e
RMSD
a
IDEN
a
Cov. BS-scor e
Lig. Name
Predicted binding site residues
1 0.07 2w16 B
0.94 0
2.32 0.345 0.98 5
1.80 PVE 59,65,67,72,643,644
2 0.06 2grxA 0.91 1
2.77 0.213 0.97 3
1.07 FTT 331,382,384
3 0.04 3rgmA 0.74 9
2.72 0.182 0.80 1
1.20 MTN 152,153,154,156,166
4 0.04 2gsk0 0.74 9
3.39 0.173 0.82 1
0.81 PEPTID E
24,34,317,325,326,389,391,43 7
5 0.04 3ddrA 0.80 3
3.56 0.146 0.89 3
0.71 HEM 449,461,514,515
(a) CscoreLB is the confidence score of predicted binding site. CscoreLB values range in between [0-1]; where a higher score indicates a more reliable ligand-binding site prediction.
(b) BS-score is a measure of local similarity (sequence & structure) between template binding site and predicted binding site in the query structure. Based on large scale benchmarking analysis, we have observed that a BS-score >1 reflects a significant local match between the predicted and template binding site.
(c) TM-score is a measure of global structural similarity between query and template protein. (d) RMSDa the RMSD between residues that are structurally aligned by TM-align.
(e) IDENa is the percentage sequence identity in the structurally aligned region.
(f) Cov. represents the coverage of global structural alignment and is equal to the number of structurally aligned residues divided by length of the query protein.
Table1. Template proteins with similar binding site.
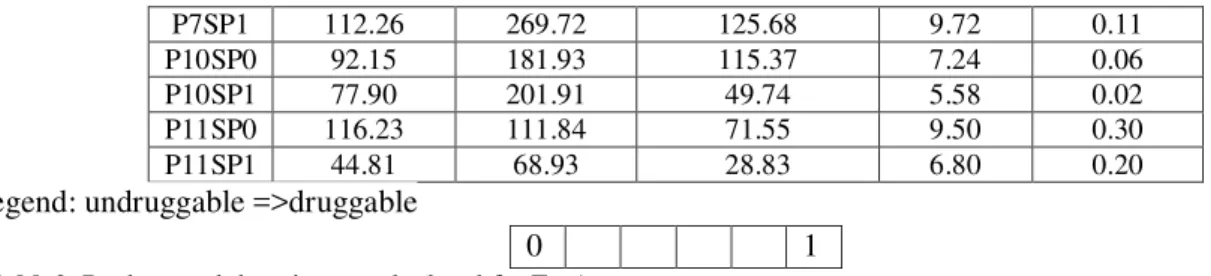
3.5. Pocket detection
Pocket descriptor table:
Name
Volume [ų] Surface [Ų] Lipo surface [Ų] Depth [Å] Drug Score
P0 2421.16 2432.22 1218.13 29.26 0.80 P1 1021.14 1057.70 583.29 17.84 0.79 P2 733.15 757.25 389.19 14.54 0.76 P3 643.07 717.43 370.42 20.12 0.82 P4 624.14 658.59 358.40 24.70 0.86 P5 524.95 533.69 254.07 18.94 0.79 P6 441.82 497.13 277.49 22.03 0.83 P7 239.93 346.89 168.76 16.74 0.62 P8 234.61 306.94 190.79 16.25 0.63 P9 182.95 191.52 100.90 13.01 0.46 P10 170.05 347.23 165.10 10.24 0.33 P11 161.04 157.98 86.56 14.55 0.53
P12 133.99 184.99 59.54 7.08 0.21
P13 128.76 294.88 138.23 7.68 0.19 P14 121.18 270.32 187.72 7.95 0.20 P15 113.61 229.46 120.40 8.81 0.24
P16 108.83 197.72 57.91 6.92 0.14
P17 101.26 300.36 177.86 8.28 0.19 Subpocket descriptor table:
Name Volume [ų] Surface [Ų] Lipo surface [Ų] Depth [Å] Drug Score
P7SP1 112.26 269.72 125.68 9.72 0.11 P10SP0 92.15 181.93 115.37 7.24 0.06 P10SP1 77.90 201.91 49.74 5.58 0.02 P11SP0 116.23 111.84 71.55 9.50 0.30 P11SP1 44.81 68.93 28.83 6.80 0.20
legend: undruggable =>druggable
0 1
Table2. Pockets and descriptors calculated for FptA
3.6. Identification of functionally and structurally important residues
Interprosurf annotated functional residues on the 3D structure of FptA.Residues Predicted by Auto Patch Analysis are:485,487,491,493,495,525,527 527,450,486,488,539,540,451,483. Results are shown in Figure 4.
Figure 4.Functional residues on FptA 3D structure.
REFRENCES:
1. Posey, J. E. &Gherardini, F. C. (2000). Lack of a role foriron in the Lyme disease pathogen. Science, 288,1651–1653
2. Ferguson, A. D. & Deisenhofer, J. (2002). TonBdependent receptors-structural perspectives.Biochim.
3. Ferguson, A. D. & Deisenhofer, J. (2004). Metal import through microbial membranes. Cell, 116, 15–24
4. He, J., Baldini, R. L., Deziel, E., Saucier, M., Zhang, Q.,Liberati, N. T. et al. (2004). The broad host rangepathogen Pseudomonas aeruginosa strain PA14carries two pathogenicityislandsharboringplantandanimalv irulencegenes.Proc.Natl Acad. Sci. USA,101, 2530–2535.
5. Poole, K. & McKay, G. A. (2003). Iron acquisition and its control in Pseudomonas
aeruginosa: many roads leadto Rome. Front. Biosci. 8, 661–686.
6. Schalk, I. J., Kyslik, P., Prome, D., Van Dorssealer, A.,Poole, K., Abdallah, M. A. &Pattus, F. (1999).Copurification of the FpvA ferric pyoverdin receptor
of Pseudomonas aeruginosa with its iron-free ligand:implication for siderophore-mediated iron transport.Biochemistry, 38, 9357–9365. 7. Schalk, I. J., Hennard, C., Dugave, C., Poole,
K.,Abdallah, M. A. &Pattus, F. (2001). Iron-frepyoverdin binds to its outer membrane receptor FpvA in Pseudomonas aeruginosa: a new mechanismfor membrane iron transport. Mol. Microbiol.39,351–360.
9. Clement, E., Mesini, P. J., Pattus, F. &Schalk, I. J.(2004). The binding mechanism of pyoverdin with theouter membrane FpvA in Pseudomonas aeruginosaisdependent on its iron-loaded status. Biochemistry, 43,7954– 7965.
10. Cobessi, D., Celia, H., Folschweiler, N., Schalk, I. J.,Abdallah, M. A. &Pattus, F. (2005). The crystalstructure of the pyoverdine outer membrane receptorFpvA from Pseudomonas aeruginosa at 3.6 angstromsresolution. J. Mol. Biol. 347, 121– 134.
11. Ankenbauer, R. G. &Quan, H. N. (1994). FptA, theFe(III)-pyochelin receptor of Pseudomonasaeruginosa:aphenolatesiderophor
e receptor homologous
tohydroxamatesiderophore receptors. J. Bacteriol. 176,307–319
12.Cox, C. D. & Graham, R. (1979). Isolation of an ironbindingcompound from Pseudomonas aeruginosa.J.Bacteriol. 137, 357–364
13.Ankenbauer, R. G., Toyokuni, T., Staley, A., Rinehart,K. L., Jr& Cox, C. D. (1988). Synthesis and biological activity of pyochelin, a siderophore of Pseudomonas aeruginosa.J.Bacteriol. 170, 5344–5351 14Namiranian, S., Richardson, D. J., Russel, D. A.
&Sodeau, J. R. (1997). Excited state properties of the siderophorepyochelin and its complex with zinc ions.
15.Visser, M. B., Majumdar, S., Hani, E. &Sokol, P. A.(2004). Importance of the ornibactin and pyochelin
siderophore transport systems in Burkholderiacenocepacia lung infections. Infect. Immunol. 72,2850–2857.
16.Hoegy, F., Celia, H., Mislin, G. L., Vincent, M., Gallay,J. &Schalk, I. J. (2005). Binding of iron-free siderophore