Moderately progressive
Ullrich congenital muscular dystrophy
Gerson Carakushansky,1 Marcia Gonçalves Ribeiro,2 Evelyn Kahn3
93 Introduction
The term congenital muscular dystrophy (CMD) refers to a heterogeneous group of inherited disorders in which muscle weakness is irst apparent at birth or in infancy.1 With the discovery of causative mutations in multiple genes in the last 2 decades, the concept of CMD has evolved from a narrowly deined clinical picture to a more inclusive group of subtypes deined by genes in which causative mutations occur. The clinical manifestations may overlap within CMD subtypes. Nonetheless, the general term CMD remains useful for pediatricians by providing a useful framework for
the diagnostic approach to the infant or young child with muscle weakness.
Establishing the speciic CMD subtype can help clarify prognosis and inheritance pattern in the involved families. To achieve this challenging task, it is necessary to rely on medical and family history; physical, neurologic and visual examinations; measurement of serum creatine kinase concentration; neuroimaging; muscle and/or skin biopsy for histological examination and immunohistochemistry; and genomic testing.
1. Doutor. Professor titular, Serviço de Genética, Instituto de Pediatria e Puericultura Martagão Gesteira (IPPMG), Universidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro, RJ, Brazil.
2. Doutora. Professora adjunta, Serviço de Genética, IPPMG, UFRJ, Rio de Janeiro, RJ, Brazil. 3. Mestre. Professora assistente, Serviço de Genética, IPPMG, UFRJ, Rio de Janeiro, RJ, Brazil.
No conflicts of interest declared concerning the publication of this article.
Suggested citation: Carakushansky G, Ribeiro MG, Kahn E. Moderately progressive Ullrich congenital muscular dystrophy. J Pediatr (Rio J). 2012;88(1):93-6.
Manuscript submitted Feb 11 2011, accepted for publication May 17 2011. http://dx.doi.org/10.2223/JPED.2112
Abstract
Objectives: To describe genetic and clinical features of Ullrich congenital muscular dystrophy (UCMD), and to report the case of a patient diagnosed with UCMD after an exhaustive investigation, which included collagen VI immunohistochemical and genomic analyses.
Description: This study was based on clinical, immunohistochemical assessment of muscle tissue and genomic analysis of dermal ibroblasts of a 7 1/2-year old boy and of the DNA of his parents. Clinical aspects and differential diagnosis with other disorders are discussed.
Comments: A better knowledge of congenital muscular dystrophies will improve the number of correct diagnoses and open new horizons for the treatment of such diseases. Genetic evaluation of UCMD patients has relevant implications for prognosis and genetic counseling of the family. The dissemination of this disorder in the pediatric community is advisable, because of the early onset of clinical manifestations and the fact that it is frequently misdiagnosed or not diagnosed at all.
J Pediatr (Rio J). 2012;88(1):93-6: Ullrich congenital muscular dystrophy, congenital muscular dystrophy, joint hyperlaxity, collagen VI.
0021-7557/12/88-01/93 Jornal de Pediatria
Copyright © by Sociedade Brasileira de Pediatria
O
94 Jornal de Pediatria - Vol. 88, No. 1, 2012
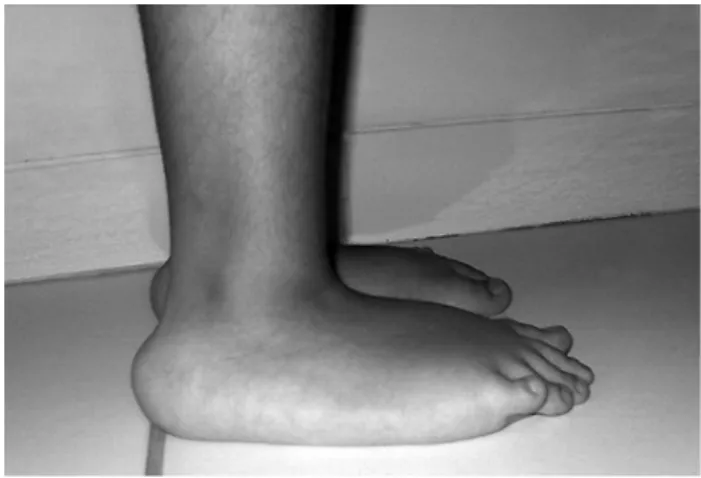
Figure 1 - Foot of the patient showing the characteristic protuberant calcaneus
Ullrich congenital muscular dystrophy - Carakushansky G et al.
Collagen VI genes may be responsible not only for Ullrich congenital muscular dystrophy (UCMD), but also for Bethlem myopathy (BM) and phenotypes intermediate to these two entities. Altogether they form a group of CMDs known as collagen VI-related myopathies.2
UCMD was originally described in 1930 by the German pediatrician Otto Ullrich3 as a disabling and life-threatening
muscular dystrophy. The author based his description on the indings in two unrelated boys with an unusual form of CMD. The clinical indings included generalized muscle weakness, contractures of the proximal joints, and hyperlexibility of the distal joints from birth or early infancy. High-arched palate, protuberant calcanei, and normal intelligence are other characteristics of the disease. However, only in the present century such phenotype has been associated with a muscular deiciency of collagen VI caused by different types of recessive and dominant mutations in the three collagen genes (COL6A1, COL6A2, and COL6A3).4,5
The true worldwide incidence of UCMD is unknown, but it is emerging as the second most common form of CMD.6 In Japan, UCMD accounts for 7.2% of CMD cases.7 There is no precise estimate for its frequency in
Brazil, probably due to the lack of molecular studies that assure a correct diagnosis in patients presenting with CMD associated with joint laxity.8
We here report a boy diagnosed with UCMD after an exhaustive investigation, which included collagen VI immunohistochemical and genomic analyses.
Case description
The patient is a Caucasian boy, currently 7 1/2 years of age, and the only child of young and healthy unrelated parents with no family history of neuromuscular diseases. He was born at term by normal vaginal delivery following an uncomplicated pregnancy with apparently normal fetal movements. The Apgar index was 8/10.
Hypotonia was present at birth. Motor milestones were delayed. The patient was able to sit at 12 months, crawl unsupported at 16 months, and walk unsupported at 24 months. At school, he was unable to run and to participate in regular physical activities with other children of the same age. His language and cognitive development were normal. The mother complained about the constant dryness and roughness of some areas of his skin.
Serum creatine kinase level was in the normal range. Electromyography revealed a chronic and inespeciic myopathic pattern of muscle discharges. A muscle biopsy was performed at the age of 3 years and 10 months, and showed a dystrophic pattern with variation in muscle iber size, moderate proliferation of endomysial and perimysial connective tissue, and variation of muscle iber diameter,
as well as scattered degenerating ibers. Results of cardiac evaluation, including echocardiography, were normal.
His weight and height plotted at the 75th percentile, whereas his head circumference plotted at the 50th percentile. The palate was arched. Skin examination revealed papular, “sandpaper” rash on the extensor surfaces of the upper arms and thighs. Abnormal hypertrophic scarring with keloid formation was present on the site of the muscle biopsy.
The patient presented a diffuse muscle weakness, pes planus, and distal hyperlaxity of ingers and toes. Posteriorly protruding calcanei were seen bilaterally (Figure 1). He was still able to walk limited distances but was not able to get up from the loor autonomously and had dificulty in climbing stairs.
Collagen VI immunostaining: frozen sections of the muscle of the patient were still available, and were simultaneously immunostained with a monoclonal antibody against collagen VI and a polyclonal antibody against laminin (L9393). In normal muscle control, collagen VI colocalized with laminin in the basement membrane surrounding muscle ibers. In the patient’s muscle, collagen VI protein was not seen in the basement membrane, even though the basement membrane appeared intact, as shown by the uniform laminin staining (Figure 2 – available online only). Abundant collagen VI protein, however, was found in the interstitial and perivascular regions between muscle ibers.
Jornal de Pediatria - Vol. 88, No. 1, 2012 95
alpha 1 chain, changing a glycine to an arginine in the triple helical domain (Gly269Arg), which was conirmed on genomic DNA. Blood of the parents was also genotyped and revealed that neither parent carries the corresponding mutation.
Discussion
Until few years ago, BM and UCMD were considered as separate entities with distinct modes of inheritance. BM is a relatively mild dominantly inherited disorder characterized by proximal weakness and distal joint contractures.2,10 However, recent advances in research on
the molecular mechanism of both diseases and on their complex genotype/phenotype correlations made it clear that BM and UCMD in fact represent opposite end points of a clinical continuum in which individuals presenting with intermediate phenotypes could be considered to have either “mild UCMD” or “severe BM.”
Abnormal motor development is constant in patients reported with UCMD, ranging from inability to run in the less-severely involved patients to complete failure to acquire gait in the severe cases. A recent study of 49 individuals with collagen VI myopathy11 referred to three phenotypes: early/severe, in which ambulation was never achieved; moderate progressive, in which ambulation was attained and lost; and mild, in which ambulation lasted into adulthood. Of importance is the fact that missense dominant de novo mutations affecting glycine were mostly observed in individuals classiied in the moderate-progressive group, which is consistent with what was found in our patient.
In 13 patients with UCMD aged 15 years or older, Nadeau et al.12 observed that decline in motor and respiratory functions was more rapid in the irst decade of life. The age at constant wheelchair dependency was variable, from 3.5 to 19 years (mean: 11.1 years).
In the neonatal period, the differential diagnosis of UCMD should include other conditions that cause early muscle weakness, such as the CMDs not related to collagen VI, congenital myopathies, and spinal muscular atrophy. The CMDs that are not collagen VI related do not generally present with the distal hyperlaxity characteristic of UCMD, and are usually associated with serum creatine kinase levels higher than those observed in UCMD. Furthermore, immunohistochemistry analysis carried out on muscle biopsy material, along with gene testing, may be of help in the diagnosis of some CMD subtypes. Spinal muscular atrophy can be ruled out with SMN1 gene testing.
The various skin changes that have been described in collagen VI-related myopathies were also present in our patient.13 Such changes usually include predisposition to
keratosis pilaris over the extensor surfaces of upper and lower limbs and abnormal hypertrophic scarring, with, for instance, the occurrence of keloids and “cigarette-paper” scars, as
the most important manifestations. In some cases, keloids can be extremely dramatic, and this issue is important not only to be considered as a diagnostic clue, but also when counseling patients ahead of surgical intervention.
Immunohistochemical assessment with double immunolabeling has proved to be a valuable guide to direct molecular analysis and substantiate the clinical diagnosis of patients under investigation for the possibility of having a collagen VI-related myopathy.14
Due to the large size of the genes involved in such myopathies, molecular diagnosis is arduous and complex. Two main mutational mechanisms underlie classical UCMD: heterozygous dominant negative mutations and recessive loss-of-function mutations.15 Gene testing is considered the gold standard for the achievement of a deinitive diagnosis, but is currently available only at a few medical centers around the world. The type of mutation found may also play an important role in the prognostic deinition of affected individuals, mainly after some genotype/ phenotype correlations have started to emerge, based on the observation of selected groups of patients.
Genetic counseling of UCMD is challenging, due to the genetic heterogeneity of this condition, being desirable that every possibility of genetic transmission should be considered in each particular situation. Consequently, the risk of recurrence for parents without a family history of the disease who delivered a single UCMD child may range from the high 25% typical risk of recessive mutations to the theoretical risk of germline mosaicism caused by de novo dominant mutations.16 Since our patient was heterozygous for a de novo dominant negative mutation and the irst child of a young healthy couple without family history of neuromuscular disease, genetic counseling will be very helpful for their future reproductive planning.
Acknowledgments
The authors are grateful to Dr. Carsten Bonnemann, from the Division of Neurology of the Children’s Hospital of Philadelphia, and to the University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA, for providing the immunohistochemical assessment of frozen muscle biopsy, the genomic analysis of the skin ibroblasts of the patient and the genotyping of his parents.
References
1. Mendell JR, Boué DR, Martin PT. The congenital muscular
dystrophies: recent advances and molecular insights. Pediatr Dev Pathol. 2006;9:427-43.
2. Bertini E, Pepe G. Collagen type VI and related disorders: Bethlem myopathy and Ullrich scleroatonic muscular dystrophy. Eur J Paediatr Neurol. 2002;6:193-8.
96 Jornal de Pediatria - Vol. 88, No. 1, 2012
Correspondence: Gerson Carakushansky
Rua General Artigas, 104/401 – Leblon CEP 22441-140 – Rio de Janeiro, RJ – Brazil Tel.: +55 (21) 9139.4680
Fax: +55 (21) 2549.2811 E-mail: [email protected] 3. Ullrich O. Kongenitale, atonisch-sklerotische Muskeldystrophie,
ein weiterer Typus der heredodegenerativen Erkrankungen des neuromuskularen Systems. Z Ges Neurol Psychiatr. 1930;126:171-201.
4. Demir E, Sabatelli P, Allamand V, Ferreiro A, Moghadaszadeh B, Makrelouf M, et al. Mutations in COL6A3 cause severe and mild phenotypes of Ullrich congenital muscular dystrophy. Am J Hum Genet. 2002;70:1446-58.
5. Baker NL, Morgelin M, Peat R, Goemans N, North KN, Bateman JF, et al. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet. 2005;14:279-93.
6. Muntoni F, Voit T. The congenital muscular dystrophies in
2004: a century of exciting progress. Neuromuscul Disord. 2004;14:635-49.
7. Okada M, Kawahara G, Noguchi S, Sugie K, Murayama K, Nonaka I, et al. Primary collagen VI deiciency is the second most
common congenital muscular dystrophy in Japan. Neurology. 2007;69:1035-42.
8. Reed UC. Congenital muscular dystrophy. Part I: a review
of phenotypical and diagnostic aspects. Arq Neuropsiquatr. 2009;67:144-68.
9. Pan TC, Zhang RZ, Sudano DG, Marie SK, Bönnemann CG, Chu ML. New molecular mechanism for Ullrich congenital muscular dystrophy: a heterozygous in-frame deletion in the COL6A1 gene causes a severe phenotype. Am J Hum Genet. 2003;73:355-69.
10. Jobsis GJ, Boers JM, Barth PG, de Visser M. Bethlem myopathy:
a slowly progressive congenital muscular dystrophy with
contractures.1999;122:649-55.
11. Briñas L, Richard P, Quijano-Roy S, Gartioux C, Ledeuil C, Lacène E, et al. Early Onset Collagen VI Myopathies: Genetic and Clinical
Correlations. Ann Neurol. 2010;68:511-20.
12. Nadeau A, Kinali M, Main M, Jimenez-Mallebrera C, Aloysius A, Clement E, et al. Natural history of Ullrich congenital muscular
dystrophy. Neurology. 2009;73:25-31.
13. Nadeau A, Muntoni F. Skin changes in Ullrich congenital muscular dystrophy.Neuromuscul Disord. 2008;18:982.
14. Jimenez-Mallebrera C, Maioli MA, Kim J, Brown SC, Feng L, Lampe AK, et al. A comparative analysis of collagen VI production in muscle, skin and ibroblasts from 14 Ullrich congenital muscular dystrophy patients with dominant and recessive COL6A mutations.
Neuromuscul Disord. 2006;16:571-82.
15. Foley AR, Hu Y, Zou Y, Yang M, Medne L, Leach M, et al. Large
genomic deletions: a novel cause of Ullrich congenital muscular
dystrophy. Ann Neurol. 2011;69:206-11.
16. Lampe AK, Zou Y, Sudano D, O’Brien KK, Hicks D, Laval SH, et al. Exon skipping mutations in collagen VI are common
and are predictive for severity and inheritance. Hum Mutat. 2008;29:809-22.