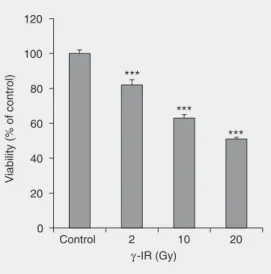
Effects of gamma-radiation on cell growth, cycle arrest, death, and superoxide dismutase expression by DU 145 human prostate cancer cells
Texto
Imagem



Documentos relacionados
To further clarify the underlying mechanism by which 2-ME induced G2/M cell cycle arrest in human UC cells, we analyzed the level of cell cycle regulatory proteins, including
The suppression of cancer cell growth has been known to be primarily mediated by apoptosis and cell cycle arrest. We next applied flow cytometry to analyze cell cycle phase
In the present study, we found that CMTM3 was frequently downregulated or silenced in testicular cancer cell lines and primary tumors (Figure 1, Table 1).We further performed
Analysis of cell cycle distribution and apoptosis in melanoma cells overexpressing NOP14 showed that NOP14 overexpression induced cell cycle arrest at the G1 phase and
Diallyl disulfide (DADS) inhibits growth and induces cell cycle G 2 /M arrest in human gastric cancer MGC803 cells.. In
STAT3 targeting shRNA can remarkably silence the expression of the gene STAT3 in HT-29 cells, resulting in inhibition of cell growth and cell cycle arrest at the G 0 /G 1 phase.
Lebein, a snake venom disintegrin, suppresses human colon cancer cells proliferation and tumor-induced angiogenesis through cell cycle arrest, apoptosis induction
In summary, results showed that aqueous HUANG-LIAN inhibited cell proliferations by inducing apoptosis and cell-cycle arrest in A549 breast cancer cells. These
