Use of Recombinant Virus Replicon Particles
for Vaccination against
Mycobacterium
ulcerans
Disease
Miriam Bolz1,2, Sarah Kerber1,2, Gert Zimmer3, Gerd Pluschke1,2*
1Swiss Tropical and Public Health Institute, Basel, Switzerland,2University of Basel, Basel, Switzerland, 3Institute of Virology and Immunology (IVI), Mittelhäusern, Switzerland
Abstract
Buruli ulcer, caused by infection withMycobacterium ulcerans, is a necrotizing disease of the skin and subcutaneous tissue, which is most prevalent in rural regions of West African countries. The majority of clinical presentations seen in patients are ulcers on limbs that can be treated by eight weeks of antibiotic therapy. Nevertheless, scarring and permanent dis-abilities occur frequently and Buruli ulcer still causes high morbidity. A vaccine against the disease is so far not available but would be of great benefit if used for prophylaxis as well as therapy. In the present study, vesicular stomatitis virus-based RNA replicon particles encoding theM.ulceransproteins MUL2232 and MUL3720 were generated and the expres-sion of the recombinant antigens characterizedin vitro. Immunisation of mice with the recombinant replicon particles elicited antibodies that reacted with the endogenous anti-gens ofM.ulceranscells. A prime-boost immunization regimen with MUL2232-recombinant replicon particles and recombinant MUL2232 protein induced a strong immune response but only slightly reduced bacterial multiplication in a mouse model ofM.ulceransinfection. We conclude that a monovalent vaccine based on the MUL2232 antigen will probably not sufficiently controlM.ulceransinfection in humans.
Author Summary
Infection withMycobacterium ulceranscan lead to a slow progressing, ulcerative disease of the skin and underlying soft tissue called Buruli ulcer. The disease is most prevalent in rural African communities with limited access to health care facilities. The most efficient means to prevent the disease, a vaccine against Buruli ulcer is not available to date. In the present study we investigated the immunogenicity and protective potential of a single cycle virus system expressing the twoM.ulceransantigens MUL2232 and MUL3720. Immunization of mice with those vesicular stomatitis virus replicon particles led to the induction of humoral as well as cellular immune responses in the immunized animals. Subsequent challenge experiments in a mouse model ofM.ulceransinfection demon-strated only a limited reduction of bacterial burden in mice immunized with a
prime-OPEN ACCESS
Citation:Bolz M, Kerber S, Zimmer G, Pluschke G (2015) Use of Recombinant Virus Replicon Particles for Vaccination againstMycobacterium ulcerans
Disease. PLoS Negl Trop Dis 9(8): e0004011. doi:10.1371/journal.pntd.0004011
Editor:Christian Johnson, Fondation Raoul Follereau, FRANCE
Received:May 19, 2015
Accepted:July 27, 2015
Published:August 14, 2015
Copyright:© 2015 Bolz et al. This is an open access article distributed under the terms of theCreative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Data Availability Statement:All relevant data are within the paper and its Supporting Information files.
Funding:The work presented here was funded by the European Community’s Seventh Framework Programme (FP7 N° 241500, Buruli Vac). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
boost approach with MUL2232. Most probably, a vaccine formulation with only one anti-gen will not be able to provide protection against Buruli ulcer in humans.
Introduction
Mycobacterium ulceranscauses Buruli ulcer (BU), a disease of the skin and underlying subcuta-neous tissue, which is reported from over 30 countries worldwide [1]. BU is most prevalent in West African countries and mainly affects children less than 15 years of age, living in remote, rural areas [2]. The natural reservoir ofM.ulceranshas not been identified so far and also the mode of transmission of this pathogen remains unclear [3].
M.ulceransproduces a macrolide exotoxin called mycolactone, which induces apoptosis in mammalian cells and leads to the typical clinical presentation of ulcerative BU skin lesions after the overlying epidermis has collapsed [4]. Non-ulcerative forms of the disease are nodules or papules, oedema and plaques [5]. BU was traditionally treated by wide surgical excision of the affected skin and skin grafting if necessary. Since 2004, treatment of patients for eight weeks with the antibiotics rifampicin and streptomycin is recommended as standard therapy by the World Health Organization (WHO) [6]. Even though the use of antibiotics has reduced recurrence rates to less than 2% [7–9], patients are often left with scars and lifelong disabilities [10]. A vaccine against BU would therefore be of high value to prevent and treat the disease [11].
As opposed toM.tuberculosisandM.leprae, which are both intracellular pathogens for which T helper 1 (TH1) cellular immune responses are essential for infection control [12],M. ulceransis found in extracellular clumps in the necrotic subcutaneous skin tissue of advanced lesions. However, there is evidence for an initial intracellular stage in macrophages during the early phase of infection [13,14]. Correlates of protection have not been identified, and in partic-ular it is not known, whether antibodies specific for surface antigens ofM.ulceranshave pro-tective activity [15].
Vaccination with Bacille Calmette-Guérin (BCG) seems to confer a transient protection from BU and a shorter duration of ulcers [16–18]. In a mouse model ofM.ulceransinfection, vaccination with either BCG or a mycolactone-negativeM.ulceransmutant strain delayed the onset of foot pad swelling [19,20]. A limited protective efficacy has also been achieved with monovalent DNA-based protein subunit vaccine formulations targeting either the mycolyl-transferase antigen (Ag) 85A fromM.bovisorM.ulcerans[21,22] or mycolactone polyketide synthase domains that are encoded on the giantM.ulceransplasmid pMUM001 [23].
In the present study we used this safe vaccine vector platform to characterize the immuno-genicity of twoM.ulceransprotein antigens. In view of the mainly extracellular nature ofM.
ulcerans, we chose antigens known to be highly expressed on the surface of the bacteria. The 18 kDa small heat shock protein (MUL2232) was identified amongst the most immunodominant antigens expressed byM.ulceranswhen serum responses from people living in endemic areas were analysed [32]. A homologue of MUL2232 was found in the genome ofM.lepraebut nei-ther inM.bovisnor inM.tuberculosis. Similar to theM.lepraehomologue, MUL2232 is associ-ated with the cell wall fraction ofM.ulcerans[32,33]. The second antigen chosen, MUL3720, is a 21 kDa protein with a putative lectin and a peptidoglycoan-binding domain that does not have homologues inM.lepraeorM.tuberculosis[34,35]. It is highly expressed byM.ulcerans
and may play a role in cell attachment and cell-cell interactions [35], making it a candidate antigen for vaccine development. We generated recombinant single-cycle VSV vectors encod-ing the selected antigens individually and analysed their expression in mammalian cell lines. We further analysed the immunogenicity of the generated VSV vectors and evaluated two short immunization protocols. Finally, we investigated the protective potential of such immu-nization schemes in an experimental mouse model ofM.ulceransinfection.
Materials and Methods
Ethics statement
All animal experiments were conducted in compliance with the Swiss animal protection law and approved by the animal welfare committee of the Canton of Basel (authorization number 2375) and the Canton of Vaud (authorization number 2657).
Cells
Baby hamster kidney 21 (BHK-21) cells were obtained from the German Cell Culture Collec-tion (DSZM, Braunschweig). Cells were grown in Earle’s minimal essential medium (MEM, Life Technologies) supplemented with 5% foetal bovine serum (FBS, Biowest). BHK-G43, a transgenic BHK-21 cell line expressing the VSV G protein in an inducible manner, was main-tained as described previously [36]. Murine L929 fibroblasts (ATCC, The Global Bioresource Center) were cultivated in RPMI medium (Gibco) supplemented with 10% foetal calf serum (FCS, Sigma), 2 mM glutamine (Gibco) and 0.05 mMβ-mercaptoethanol (Gibco).
Generation of recombinant vesicular stomatitis virus replicon particles
expressing
M
.
ulcerans
codon optimized antigens
The potential protein vaccine candidate antigens MUL_2232 (GenBank accession number 4550596) and MUL_3720 (GenBank accession number 4553013) ofM.ulceransAgy99 were ordered as codon optimized genes for expression in humans (GenScript) and received in pUC57 plasmids. For generation of recombinant VSV replicon particles (VRPs), codon opti-mized target antigens were amplified by PCR and inserted into the pVSVΔ
G plasmid using single MluI and BstEII restriction sites upstream and downstream of the fourth transcription unit, replacing the VSV G gene [37]. Sequence integrity of the resulting plasmids was con-firmed by Sanger sequencing. Recombinant VRPs were generated as described previously [38]. In brief, BHK-G43 cells were infected with recombinant MVA-T7 virus expressing T7 RNA polymerase [39]. Subsequently, the infected cells were transfected with plasmids driving T7 RNA polymerase-mediated expression of the VSV proteins N, P, and L, and with pVSVΔ
G (MUL2232) or pVSVΔG(MUL3720) driving T7 RNA polymerase-mediated transcription of
adding mifepristone (Sigma) to the cell culture medium. At 24 hours post transfection cells were detached with trypsin and seeded along with an equal number of fresh BHK-G43 cells into T-75 flasks. Cells were then incubated at 37°C for another 24 hours in the presence of mifepristone. Cell culture supernatant was clarified by low-speed centrifugation and by passage through a 0.2μm pore filter. The VRPs in the clarified cell culture supernatant were further
propagated on mifepristone-induced BHK-G43 cells and stored at -70°C. VRPs were titrated on BHK-21 cells taking advantage of the eGFP reporter protein.
Western blot analysis
Detection of vaccine antigens in VRP-infected cells. Equal numbers of confluent L929 fibroblasts were infected with VRPs at a multiplicity of infection (MOI) of 10 for 6 hours at 37°C. Cells were washed with ice-cold phosphate buffered saline (PBS; Sigma) and harvested for lysis. Protein lysates of both the soluble and insoluble fraction of the cells were produced with a cell fractionation kit (Abcam) according to the manufacturer’s instructions. Equal amounts of protein lysates as well asM.ulceranslysate in an appropriate dilution were resolved on prefabricated 4–12% gradient gels (NuPAGE Novex 4–12% Bis-Tris Gel; Invitrogen) with MES running buffer (Invitrogen) according to the manufacturer’s directions. A dry-blotting system (iBlot; Invitrogen) was used to transfer proteins to nitrocellulose membranes. Mem-branes were blocked and specific proteins detected with in house MUL2232 or anti-MUL3720 mouse mAbs followed by HRP-conjugated goat anti-mouse IgGγ-chain mAb (Southern Biotech, 1030–05). Blots were developed using ECL Western blotting detection reagents (ECL Western blotting Substrate; Pierce).
Cross-reactivity of the elicited antibodies in mouse sera with the native antigen inM. ulceranslysate. 10μg ofM.ulceranswhole cell lysate was resolved on a one well prefabricated
4–12% gradient gel (NuPAGE Novex 4–12% Bis-Tris Gel; Invitrogen) with MES running buffer according to the manufacturer’s instructions. The blocked membrane was subsequently cut into thin strips that were individually incubated with appropriate dilutions of serum of immunized mice for 2 hours. Secondary antibody and development were performed as described above.
Immunofluorescence analyses
For indirect immunofluorescence analysis, BHK-21 cells were grown on 12 mm diameter cover slips (2 x 105cells/well) and infected with VRPs (106infectious units/well) for 6 hours at 37°C. Cells were fixed with 3% paraformaldehyde (PFA) and washed with PBS containing 0.1 M of glycine. The cells were permeabilized with 0.25% (v/v) Triton X-100 and subsequently incu-bated for 1 hour with anti-MUL2232 or anti-MUL3720 mAbs in appropriate dilutions in 1% bovine serum albumin (Sigma). For detection of antigen-bound primary antibodies, cells were incubated with an Alexa Flour 546 labelled anti-mouse IgG secondary antibody (1/500; Molec-ular Probes, A-11018). The cells were washed with distilled water, embedded in Mowiol 4–88 (Sigma) mounting medium, and analyzed with a Leica TCS SL confocal microscope and LCS software (Leica Microsystems AG, Glattbrugg, Switzerland).
ELISA on recombinant protein
product was measured at 405 nm with a microplate reader (Sunrise Absorbance Reader; Tecan). The threshold for endpoint titer determination was defined as the double of the mean measurements plus the mean standard deviation of a dilution series done without primary anti-body and a dilution series done with pre-bleed serum. Individual serum dilution series were approximated with sigmoidal dose-response curves and the reciprocal dilution of the intersec-tion between the curve and the threshold was defined as individual endpoint titer.
Mouse immunization studies
All animal studies were conducted in 8 weeks old female BALB/c mice (Janvier). VRPs (107 fluorescence-forming units) were either applied subcutanously (s.c.) in the neck (for evaluation of the vaccination protocol) or intramuscularly (i.m.) into the right caudal tight muscle using a volume of 100μl or 30μl, respectively. When several immunizations were conducted,
injec-tions were performed in three week intervals. For prime-boost vaccination regimen, 30μg of
non-adjuvanted recombinant protein were applied s.c. into the neck. Blood was collected from the tail vein prior to every immunization as well as 5/6 and 14 days after the protein boost. Serum was prepared by centrifugation of the blood in SST Microtainer tubes (Becton, Dickin-son and Company).
Analysis of cellular immune responses
Immunized mice were euthanized 3 weeks after the second immunization. Heart blood was collected and spleens were aseptically removed and homogenized by passing through a 70μm
cell strainer (BD Falcon). Cells were then pelleted and red blood cells lysed by incubating the pellet in red blood cell lysing buffer (Sigma) for 1 minute. Remaining cells were washed several times with stemline T-cell expansion medium (Sigma) supplemented with 4mM L-Glutamine (Gibco), 1% Pen-Strep (Gibco), 2.5 ug/ml Amphotericin B (Sigma) and 0.05 mMβ -mercap-toethanol (Gibco) and finally adjusted to 4 x 106white blood cells/ml. 7.2 x 105cells per well were incubated in round-bottom microwell plates (BD Falcon) in a humidified CO2incubator
and stimulated with Concanavalin A (2μg/ml, Sigma) as positive control or recombinant
pro-teins at 5μg/ml. Supernatants were harvested after 24h for Interleukin 2 (IL-2) and 96h for
Interleukin 10 (IL-10) and Interferon gamma (IFNγ) assays and stored frozen at -20°C until analysis for the selected cytokines. Amount of cytokines in supernatants was determined with Quantikine ELISA kits for IL-2, IL-10 and IFNγ(R&D Systems).
Challenge infection experiments
TheM.ulceransstrain (S1013) used for the experimental infection of mice was isolated in 2010 from the ulcerative lesion of a Cameroonian BU patient [2]. Bacteria were cultivated for 6 weeks in Bac/T medium (Biomerieux), recovered by centrifugation, and suspended in sterile PBS to 125 mg/ml wet weight corresponding to 2.8 x 105CFU/ml as determined by plating serial dilutions on 7H9 agar plates (Difco). Three weeks after the last immunization, mice were infected with 30μl ofM.ulceranssuspension (1/100 of the stock solution in PBS) into the left
Colony forming unit (CFU) plating and enumeration of bacterial load by
real-time PCR (qPCR)
Mouse feet designated for enumeration ofM.ulceransbacteria were immediately removed above the ankle after euthanasia, shortly dipped into 70% ethanol, then dried under the laminar flow, cut in 4 pieces with a scalpel and transferred to 750μl of Bac/T medium in reinforced
hard tissue grinding tubes (MK28-R, Precellys). Tissue homogenization was performed with a Precellys 24-Dual tissue homogenizer (3 x 20 s at 5000 rpm with 30 s break), the lysate was transferred to a new tube and the lysis tube still containing tissue remains was refilled with 750μl of Bac/T medium. The remains were homogenized a second time and the two lysates
pooled.
CFU plating. 250μl of the foot pad lysate was decontaminated with 0.5 M NaOH as
described previously [41]. The pellet of decontaminated lysate was dissolved in 500μl of Bac/T
medium and appropriate dilution series plated on 7H9 agar plates. Following 150 days of incu-bation at 30°C colonies were counted and the concentration of bacteria expressed as CFU.
DNA extraction and qPCR. DNA from 100μl of a 1 to 50 dilution of the foot pad lysate
in PBS was extracted as described by Lavender and Fyfe [42]. Extracted DNA was analysed for IS2404 by qPCR as previously described [42]. Ct values were converted into genome copy numbers per foot pad by applying the standard curve established for IS2040 byFyfe et al. [43].
Histopathology
Mouse feet designated for histopathological analysis were removed above the ankle and imme-diately transferred to 10% neutral-buffered Formalin solution (approx. 4% formaldehyde, Sigma) for fixation during 24 hours at room temperature. Subsequently, the feet were decalci-fied in 0.6 M EDTA and 0.25 M citric acid for 12 days at 37°C and transferred to 70% ethanol for storage and transport. The samples were dehydrated and embedded into paraffin. 5μm
thin sections were cut, deparaffinised, rehydrated, and stained with Haematoxylin/Eosin (HE, Sigma, J.T. Baker) or Ziehl-Neelsen/Methylene blue (ZN, Sigma) according to WHO standard protocols [44]. Stained sections were mounted with Eukitt mounting medium (Fluka). Pictures were taken with a Leica DM2500B microscope or with an Aperio scanner.
Statistical analysis and image processing
Differences of bacterial load in infected foot pads were statistically analysed by the Mann-Whitney test using Graph Pad Prism (Version 6.03). Results of cytokine production were sub-jected to log10transformation and subsequently analysed with SAS software (SAS Institute,
Cary, USA, release 9.3) using linear mixed models adjusted for random effects. Image process-ing and picture panel assembly was performed with Photoshop software (Adobe Photoshop CS6 Extended, version 13.0.1).
Results
Expression of the
M
.
ulcerans
vaccine candidate antigens MUL2232 and
MUL3720 by cells infected with recombinant virus replicon particles
Candidate vaccines were generated by replacing the VSV glycoprotein G gene with theM.
ulceransgenes MUL2232 or MUL3720 (Fig 1A). In order to ease virus detection and titration, the coding sequence for the enhanced green fluorescent protein (eGFP) was inserted as an additional transcription cassette downstream of theM.ulceransgenes. The recombinant viruses VSVΔG(MUL2232) and VSVΔG(MUL3720) as well as the control virus VSVΔG,
produced and propagated in the helper cell line BHK-G43 providing the VSV G proteinin trans. As expected from other studies with similar constructs, the trans-complemented parti-cles were able to infect a variety of different mammalian cell lines but were unable to release progeny viruses [27,45]. We thus refer to them as to virus replicon particles (VRPs).
In order to study the expression of MUL2232 and MUL3720 in infected cells, BHK-21 cells were infected with the three different VRPs generated and analysed by using MUL2232 and MUL3720-specific mouse monoclonal antibodies (mAbs) in immunofluorescence microscopy. Both antigens accumulated in the cytosol of the infected BHK-21 cells (Fig 1B1 and 1B2). While in the immunofluorescence analysis MUL2232 appeared to be expressed at lower levels than MUL3720, staining intensities in Western blotting analyses were comparable for both proteins (Fig 1C). MUL2232 was mainly detected in the soluble fraction of infected L929 fibro-blasts and to a smaller part in the insoluble fraction (Fig 1C1) while MUL3720 was found in the soluble fraction only (Fig 1C2). Both proteins co-migrated with the corresponding proteins inM.ulceranslysate according to the predicted molecular mass (Fig 1C).
Immunization of mice with virus replicon particles induces
M
.
ulcerans
cross-reactive antibody responses
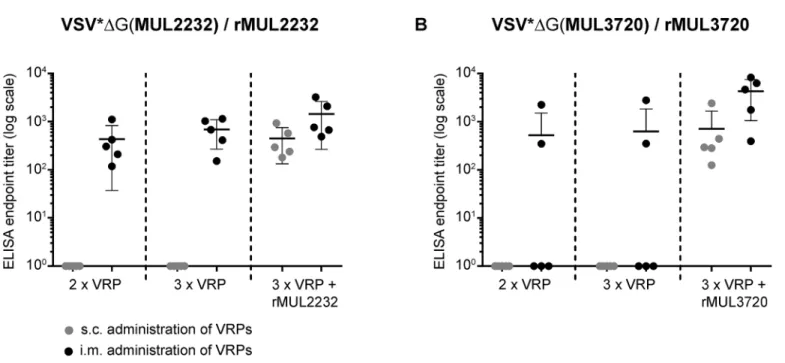
To characterize the immune responses elicited by the recombinant VRPs, we compared differ-ent immunization regimens in BALB/c mice. Humoral immune responses were assessed by ELISA on immobilized recombinant protein. Sub-cutaneous (s.c.) administration of 107VRPs did not lead to measureable antibody responses against the target antigens (Fig 2A and 2B). However, when VSVΔ
G(MUL2232) was given i.m., Immunoglobulin (Ig) G antibodies were elicited in all immunized animals. In contrast, only some mice produced antibodies following i. m. immunization with VSVΔ
G(MUL3720) (Fig 2A and 2B). Therefore induction of antibod-ies solely by i.m. immunization with VSVΔG(MUL3720) was no longer pursued in subsequent
experiments. Independently of the route of administration, both VRPs primed the immune system, as demonstrated by the fast humoral immune response to an adjuvant-free booster immunization (s.c.) with 30μg of the corresponding recombinant protein (Fig 2A and 2B). Six
days after the booster injection, antibody titers were generally higher in mice primed i.m. than in mice primed s.c. (Fig 2A and 2B). Therefore, only the i.m. route was employed for VRP administration in subsequent experiments. In addition, a dose of 107VRPs per immunization was generally used.
In a next step, we assessed the potential of a shorter prime-boost immunization strategy by immunizing the animals only once i.m. with VSVΔ
G(MUL2232) or VSVΔ
G(MUL3720) and three weeks later with 30μg of rMUL2232 or rMUL3720 via the subcutaneous route in the
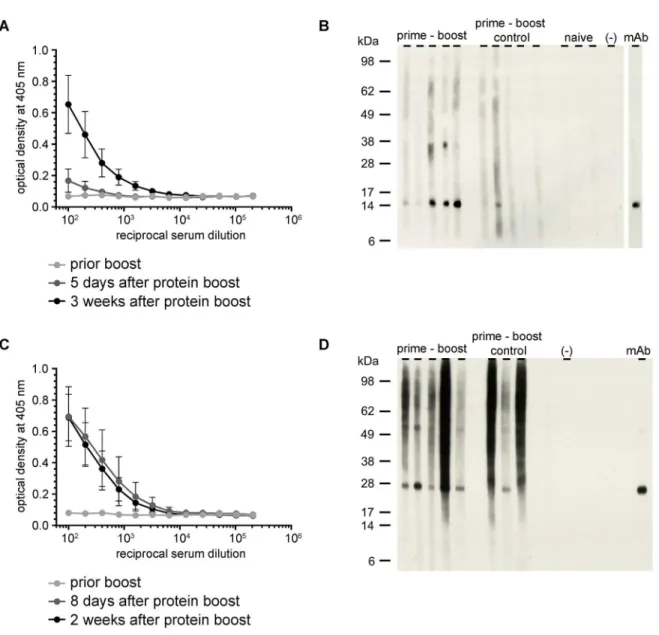
absence of adjuvant. Five days after the rMUL2232 boost, the ELISA IgG titers were only mar-ginally higher than those observed prior to the boost, but increased further in the subsequent two weeks (Fig 3A). On the other hand, administration of rMUL3720 led to high antibody titers already eight days after the boost (Fig 3C). Importantly, the elicited antibodies were not only cross-reactive with the recombinant proteins produced inE.colibut also with the target
Fig 1. Expression ofM.ulceransproteins by VRP-infected cells.(A) Genome maps of recombinant VSV: The genome of VSV encodes for the nucleoprotein N, phosphoprotein P, matrix protein M, glycoprotein G, and the large RNA polymerase L. In VSV*ΔG, the glycoprotein G coding region was replaced by the coding sequence for eGFP. In VSV*ΔG(MUL22332) and VSV*ΔG(MUL3720), the VSV G gene was replaced by theM.ulceransgenes MUL2232 and MUL3720, respectively. The reporter protein eGFP is encoded by an additional transcription cassette located downstream of theM.ulcerans
genes. (B) BHK-21 cells were infected with the indicated replicons and indirect immunofluorescence analysis performed with mAbs specific for MUL2232 (B1) or MUL3720 (B2) (red fluorescence). The infection of cells with VRPs is indicated by eGFP expression (green fluorescence). Scale bar equals 30μm. (C) Equal numbers of L929 fibroblasts were infected with the indicated VRPs at a MOI of 10. At 6 hours post infection, the cells were harvested and protein lysates of the soluble (s) and insoluble (i) fraction analysed by Western blotting for expression ofM.ulceransantigens using mAbs specific for MUL2232 (C1) and MUL3720 (C2), respectively.
proteins expressed byM.ulcerans, as demonstrated by Western blotting onM.ulceranslysate (Fig 3B and 3D).
To evaluate whether this immunization regimen would also elicit antigen-specific cellular immune responses, antigen-specific cytokine secretion of spleen cells was studiedin vitroafter primary immunization of mice with VSVΔG(MUL2232) and a second immunization with
either rMUL2232 or VSVΔ
G(MUL2232). Three weeks after the second immunization, spleen cells were stimulated with either rMUL2232 or the unrelated rMUL3720 as control. Stimula-tion with Concanavalin A (ConA) served as positive control for confirming the viability of the cultured spleen cells. The stimulated cell culture supernatants were analysed by ELISA for the production of the TH1 cytokine Interferon gamma (IFNγ) (Fig 4A), the pleiotropic [46]
cyto-kine Interleukin 2 (IL-2) (Fig 4B) and the TH2 cytokine Interleukin 10 (IL-10) [47] (Fig 4C).
Overall, no marked difference in terms of cytokine production was observed between the two immunization schedules. In both cases all three cytokines were produced in significantly higher amounts when cultured spleen cells were stimulated with the corresponding rMUL2232 anti-gen. In contrast, stimulation with the unrelated antigen rMUL3720 or mock stimulation had no significant effect (Fig 4). The most pronounced response was found for IL-10 (Fig 4C), indi-cating a slight polarization towards a TH2 type response. The same experimental setup with
VSVΔ
G(MUL3720) was not successful due to technical problems.
Vaccination has no marked inhibitory effect on bacterial proliferation in
an experimental BU mouse model
In a last step, we explored the protective efficacy of the VRP-based immunization in an experi-mentalM.ulceransinfection model. Groups of six BALB/c mice were immunized according to the VSVΔG(MUL2232)/rMUL2232 prime-boost regimen or two times i.m. with VSVΔG
(MUL2232). Respective control groups were immunized either with the control VRP VSVΔ
G
Fig 2. Comparison of immunization routes.Groups of five female BALB/c mice were sequentially immunized with VSV*ΔG(MUL2232) (A) or VSV*Δ
(MUL3720) (B) via the sub-cutaneous (grey dots) or intra-muscular (black dots) route. Prior to every new immunization, blood was taken and serum analysed by ELISA on the corresponding recombinant proteins (rMUL2232 or rMUL3720). After the third immunization, all mice were boosted (s.c) with the VRP correspondent, non-adjuvanted recombinant protein. Endpoint total IgG titers were determined for individual animals in one single ELISA. Mean values (line) ±standard deviations are shown.
and an rMUL2232 boost (prime-boost control) or twice with VSVΔ
G (Fig 5). Three weeks after the last immunization, the left hind foot pad of the mice was infected with 8.4 x 103M.
ulceransbacilli. The slowly progressing infection was followed by weekly measurements of the thickness of the infected foot pads with a caliper (Fig 5A). To determine the bacterial multipli-cation, mice were euthanized at day 60 after infection and foot pads either processed for histo-pathological analysis or lysed for enumeration ofM.ulceransby standard CFU plating. Quantification was additionally performed by an adapted qPCR method, suitable for the detec-tion of bacterial proliferadetec-tion in mouse foot pads over the course of the infecdetec-tion (Fig 5C).
No differences in the kinetics of footpad swelling were observed between immunized mice and the respective control animals (Fig 5A). However, 60 days post infection the number of
Fig 3. Induction of antigen-specific antibodies by a short prime-boost immunization protocol.Female BALB/c mice (n = 5) were immunized once with VSV*ΔG(MUL2232) or VSV*ΔG(MUL3720) and boosted with rMUL2232 or rMUL3720 three weeks later. Control animals (n = 5 and n = 3) were immunized with empty replicon prior to recombinant protein boost three weeks later. (A) Analysis of serially diluted immune sera by ELISA on immobilized rMUL2232. Mean values (dot)±standard deviations are shown. (B) Western blotting analysis onM.ulceranslysate using the indicated immune sera taken three weeks after protein boost. (C) Analysis of serially diluted immune sera by ELISA on immobilized rMUL3720. Mean values (dot)±standard deviation are shown. (D) Western blotting analysis onM.ulceranslysate using the indicated immune sera taken two weeks after protein boost.
CFU per foot pad was slightly but significantly lower in animals immunized with the VRP prime-protein boost regimen as compared to the prime-boost control immunized animals (Fig 5B). Quantification ofM.ulceransDNA in the footpads by insertion sequence (IS) 2404 spe-cific qPCR yielded similar results, i.e. a slight but significant reduction in bacterial multiplica-tion caused by the VRP prime-protein boost immunizamultiplica-tion, but not by two subsequent immunization with VRPs (Fig 5B and 5D). The same experiment with MUL3720 VRPs did not result in any difference between immunized and control groups (S1 Fig).
A histopathological analysis of representative foot pads confirmed the findings of a slight reduction of bacterial burden in the VSV prime-protein boost immunized animals as com-pared to the prime-boost control animals. The non-infected right foot pads served as control and appeared completely normal (Fig 6A1) with intact muscle tissue (Fig 6A2) and no appar-ent oedematous changes (Fig 6A3). In comparison, the infected left foot pads of control mice showed typical histopathological signs of BU with strong oedema (Fig 6B1 and 6B4), necrotic sole of foot (Fig 6B2), and inflammatory infiltration and extensive haemorrhages all over the foot pad (Fig 6B3). Ziehl-Neelsen/Methylene blue (ZN) staining revealed large clumps of acid fast bacilli (AFB) (Fig 6C1), not only located where they were initially injected, but also more towards the heel of the foot (Fig 6C2) and in the oedematous tissue in the upper part of the foot pad (Fig 6C5). AFB were associated with remains of infiltrating immune cells (Fig 6C3) and were also found as fibrous structures in completely necrotic tissue (Fig 6C4). In animals that received a VRP prime-protein boost immunization, a trend towards a reduction in the number of AFB clusters was observed (Fig 6D1). Furthermore, AFB appeared to be more often in close contact with infiltrating cells or were found intracellular (Fig 6D3).
Discussion
Despite substantial control efforts and improvements in treatment and diagnosis, the socioeco-nomic impact of BU on affected communities remains high. Long treatment regimens with daily i.m. injections in rural settings of West Africa, late reporting to health facilities, scarring and resulting permanent disabilities create high morbidity that could be prevented by a vaccine againstM.ulcerans. Although no such vaccine is available at the moment, reports on self-heal-ing in patients with early stages of the disease [48,49] as well as sero-epidemiological studies in BU-endemic countries [32,50] indicate that the human organism is in principle capable of inducing a protective immune responses against BU. Furthermore, protein subunit vaccination approaches have demonstrated partial protection in a BU mouse infection model [21,22,51].
In the present study, we used a viral replicon particle system for deliveringM.ulcerans pro-tein antigens to the immune system. Immunization with a high number of VRPs inducedM.
ulceransspecific antibody responses with i.m. application being superior to s.c. application. Our VRP prime-recombinant protein boost regimen not only induced a humoral but also a cel-lular immune response. This is in line with previous work showing that VRPs are able to trigger humoral as well as cellular immune responses [52].
after the final immunization, mice were euthanized. Spleen cells were taken into culture and stimulated with either Concanavalin A (ConA), rMUL2232, rMUL3720 or left unstimulated (medium). Levels of secreted cytokines were determined by commercially available ELISA kits at 24 hours (IL-2, B) and 96 hours (IFNγ, A; IL-10, C) after stimulation. Mean values (bar)±standard deviations for 5 mice tested are shown and expressed as log10(pg/ml).*p0.05;**p0.01;***p0.001;****p0.0001 (p–value adjusted for random effects in a linear mixed model). Ns equals non-significance. # Due to high non-specific rMUL3720 stimulation presumably caused by contaminants in the recombinant protein preparation, values were normalized to those of a non-immunized group of mice and z-values are depicted for all stimulations except ConA.
The bacterial proteins MUL2232 and MUL3720 were chosen as vaccine antigens because previous work suggested that these antigens are not only highly immunogenic but also associ-ated with the outer surface ofM.ulcerans[32,34,35]. Therefore, the strong humoral immune response observed following immunization of mice was expected to allow antibody-mediated opsonization of the bacteria with consequent enhanced phagocytosis, complement activation,
Fig 5. Infection of immunized mice withM.ulcerans.Groups of six immunized, female BALB/c mice were infected by s.c. injection ofM.ulcerans
suspension (30μl) into the left hind foot pad. (A) Development of the infection was followed by weekly measurements of the foot pad thickness with a caliper. The mean foot pad thickness (dot)±standard deviation is shown for each animal group. (B) At day 60 after infection, mice were sacrificed and the number of
M.ulceransbacilli in foot pads determined by classical CFU plating. (C) Quantification ofM.ulceransin foot pad lysates by IS2404 specific qPCR. Genome equivalents are shown. (D) Determination ofM.ulceransgenome equivalents in immunized and infected mice.*p0.05 (Mann-Whitney test).
or antibody-dependent cellular cytotoxicity. While we did not observe vaccination-induced reductions in foot pad swelling in our murineM.ulceransinfection model, assessment of the
Fig 6. Histopathological analysis of mouse foot pads following infection withM.ulcerans.Histological sections of foot pads fromM.ulcerans-infected mice were stained with Haematoxylin/Eosin (A1-A3, B1-B4) or Ziehl-Neelsen/Methylene blue (ZN) (C1-C5, D1-D3). The right, non-infected foot pad showed no swelling (A1), intact muscle tissue (A2) and absence of oedema in the upper part of the foot (A3). As opposed to this, the left foot of a mouse immunized with VSV*ΔG/rMUL2232 (prime-boost control) was strongly swollen 60 days post infection (B1). Cellular infiltration and beginning necrosis of the tissue was explicitly strong at the base of the foot pad, where bacteria were injected (B2) and muscle fibres started to become destroyed (B3). The upper part of the foot is highly oedematous (B4). Staining of the same foot pad for AFB revealed large amounts of bacteria in close contact to infiltrating immune cells at the base of the foot (C3). Appearance of AFB in the destroyed muscle tissue (C4). Big clumps of bacteria were also found towards the ankle of the foot (C2) and in the oedematous tissue in the upper half of the foot pad (C5). The foot pad of a mouse immunized with VSV*ΔG(MUL2232)/rMUL2232 (prime-boost) contained large amounts of AFB (D1) which were less spread in the whole foot pad. Intracellular AFB were frequently observed (D3).
bacterial load by qPCR as well as CFU plating revealed a slight, but significant reduction in bac-terial multiplication in VRP prime-protein boost immunized mice. While measurement of foot pad swelling over time is a parameter that can be followed without euthanizing mice [53,54], our results illustrate that foot pad swelling not necessarily reflects the extent of bacterial proliferation.
Despite a slight inhibitory effect on bacterial proliferation, immunization with only one tar-get antigen in a VRP prime-protein boost regimen was not sufficient to confer full protection against the experimental infection. Several factors, like the choice or number of antigens included into the VRPs, could have let to this negative result. Since it is not known, which immune effector functions are relevant for protection againstM.ulceransdisease [15], it is not clear whether lack of a strong TH1-polarisation is of major relevance for the failure to achieve a
strong protective efficacy with the immunization regimens tested. One of the advantages of the replicon system lies in the ability of VSV to tolerate incorporation of long stretches of foreign DNA into its genome [25,26]. As a modular system, it offers the possibility to design a multiva-lent subunit vaccine by combining severalM.ulceransproteins in one replicon. Furthermore the versatility of the system allows to engineer the location of the expressed protein in the infected cell, or to target specific immune cells by including genes for co-stimulatory molecules or receptors to be expressed on the surface of the infected cells [25]. Here we have demon-strated that RNA replicon particles are a very good delivery system for mycobacterial antigens, which is in particular encouraging future development of VRP-based multivalent subunit vaccines.
Supporting Information
S1 Fig. Infection of immunized mice withM.ulcerans(MUL3720 immunizations).Groups of six immunized, female BALB/c mice were infected into the left hind foot pad with 30μl of
M.ulceranssuspension (s.c.). (A) Development of the infection was followed by weekly mea-sures of the foot pad thickness with a caliper. Depicted is the mean foot pad thickness (dot) ± standard deviation of the individual differently immunized groups. (B) At day 60 after infec-tion, mice were sacrificed and the number ofM.ulceransbacilli in foot pads determined by classical CFU plating. (C) Determination ofM.ulceransgenome equivalents in immunized and infected mice.
(PDF)
Acknowledgments
We thank Martin Bratschi for providing CameroonianM.ulceransfor the mouse infection experiments and Diana Diaz and Jean-Pierre Dangy for providing monocolonal antibodies. We further thank Dr. Masato Murakami, Vincent Romanet, Caroline Stork, Ernesta Dam-massa and Patricia Barzaghi Rinaudo from Novartis Basel for excellent technical support and providing access to lab equipment for histopathology. We also thank Peter Schmid for the Aperio scans of the tissue and are very grateful for statistical support by Letizia Grize.
Author Contributions
References
1. Van der Werf TS, Van der Graaf WT, Tappero JW, Asiedu K. Mycobacterium ulcerans infection. The Lancet. 1999; 354: 1013–1018. doi:10.1016/S0140-6736(99)01156-3
2. Bratschi MW, Bolz M, Minyem JC, Grize L, Wantong FG, Kerber S, et al. Geographic distribution, age pattern and sites of lesions in a cohort of Buruli ulcer patients from the Mapé Basin of Cameroon. PLoS Negl Trop Dis. 2013; 7: e2252. doi:10.1371/journal.pntd.0002252PMID:23785529
3. Merritt RW, Walker ED, Small PLC, Wallace JR, Johnson PDR, Benbow ME, et al. Ecology and trans-mission of Buruli ulcer disease: a systematic review. PLoS Negl Trop Dis. 2010; 4: e911. doi:10.1371/ journal.pntd.0000911PMID:21179505
4. George KM, Chatterjee D, Gunawardana G, Welty D, Hayman J, Lee R, et al. Mycolactone: a polyke-tide toxin from Mycobacterium ulcerans required for virulence. Science. 1999; 283: 854–857. PMID: 9933171
5. WHO | Laboratory diagnosis of buruli ulcer. In: WHO [Internet]. [cited 2 Jun 2014]. Available:http:// www.who.int/buruli/laboratory_diagnosis/en/
6. WHO | Provisional guidance on the role of specific antibiotics in the management ofMycobacterium ulceransdisease (Buruli ulcer). In: WHO [Internet]. [cited 19 Jun 2014]. Available: zotero://attachment/ 245/
7. Chauty A, Ardant M-F, Adeye A, Euverte H, Guédénon A, Johnson C, et al. Promising clinical efficacy of streptomycin-rifampin combination for treatment of buruli ulcer (Mycobacterium ulcerans disease). Antimicrob Agents Chemother. 2007; 51: 4029–4035. doi:10.1128/AAC.00175-07PMID:17526760 8. Nienhuis WA, Stienstra Y, Thompson WA, Awuah PC, Abass KM, Tuah W, et al. Antimicrobial
treat-ment for early, limited Mycobacterium ulcerans infection: a randomised controlled trial. Lancet. 2010; 375: 664–672. doi:10.1016/S0140-6736(09)61962-0PMID:20137805
9. Sarfo FS, Phillips R, Asiedu K, Ampadu E, Bobi N, Adentwe E, et al. Clinical efficacy of combination of rifampin and streptomycin for treatment of Mycobacterium ulcerans disease. Antimicrob Agents Che-mother. 2010; 54: 3678–3685. doi:10.1128/AAC.00299-10PMID:20566765
10. Agbenorku P, Donwi IK, Kuadzi P, Saunderson P. Buruli ulcer: treatment challenges at three centres in ghana. J Trop Med. 2012; 2012: 371915. doi:10.1155/2012/371915PMID:23251191
11. Einarsdottir T, Huygen K. Buruli ulcer. Hum Vaccin. 2011; 7: 1198–1203. doi:10.4161/hv.7.11.17751 PMID:22048117
12. Prezzemolo T, Guggino G, La Manna MP, Di Liberto D, Dieli F, Caccamo N. Functional Signatures of Human CD4 and CD8 T Cell Responses to Mycobacterium tuberculosis. Front Immunol. 2014; 5: 180. doi:10.3389/fimmu.2014.00180PMID:24795723
13. Coutanceau E, Marsollier L, Brosch R, Perret E, Goossens P, Tanguy M, et al. Modulation of the host immune response by a transient intracellular stage of Mycobacterium ulcerans: the contribution of endogenous mycolactone toxin. Cell Microbiol. 2005; 7: 1187–1196. doi:10.1111/j.1462-5822.2005. 00546.xPMID:16008585
14. Torrado E, Fraga AG, Castro AG, Stragier P, Meyers WM, Portaels F, et al. Evidence for an intrama-crophage growth phase of Mycobacterium ulcerans. Infect Immun. 2007; 75: 977–987. doi:10.1128/ IAI.00889-06PMID:17145944
15. Schütte D, Pluschke G. Immunosuppression and treatment-associated inflammatory response in patients with Mycobacterium ulcerans infection (Buruli ulcer). Expert Opin Biol Ther. 2009; 9: 187–200. doi:10.1517/14712590802631854PMID:19236249
16. Nackers F, Dramaix M, Johnson RC, Zinsou C, Robert A, Biurrun DE Bakedano E, et al. BCG vaccine effectiveness against Buruli ulcer: a case-control study in Benin. Am J Trop Med Hyg. 2006; 75: 768–
774. PMID:17038709
17. Smith PG, Revill WD, Lukwago E, Rykushin YP. The protective effect of BCG against Mycobacterium ulcerans disease: a controlled trial in an endemic area of Uganda. Trans R Soc Trop Med Hyg. 1976; 70: 449–457. PMID:841647
18. BCG vaccination against mycobacterium ulcerans infection (Buruli ulcer). First results of a trial in Uganda. Lancet. 1969; 1: 111–115. PMID:4178240
19. Converse PJ, Almeida DV, Nuermberger EL, Grosset JH. BCG-mediated protection against Mycobac-terium ulcerans infection in the mouse. PLoS Negl Trop Dis. 2011; 5: e985. doi:10.1371/journal.pntd. 0000985PMID:21423646
21. Tanghe A, Content J, Van Vooren J-P, Portaels F, Huygen K. Protective Efficacy of a DNA Vaccine Encoding Antigen 85A from Mycobacterium bovis BCG against Buruli Ulcer. Infect Immun. 2001; 69: 5403–5411. doi:10.1128/IAI.69.9.5403–5411.2001PMID:11500410
22. Tanghe A, Dangy J-P, Pluschke G, Huygen K. Improved protective efficacy of a species-specific DNA vaccine encoding mycolyl-transferase Ag85A from Mycobacterium ulcerans by homologous protein boosting. PLoS Negl Trop Dis. 2008; 2: e199. doi:10.1371/journal.pntd.0000199PMID:18350112 23. Roupie V, Pidot SJ, Einarsdottir T, Van Den Poel C, Jurion F, Stinear TP, et al. Analysis of the vaccine
potential of plasmid DNA encoding nine mycolactone polyketide synthase domains in Mycobacterium ulcerans infected mice. PLoS Negl Trop Dis. 2014; 8: e2604. doi:10.1371/journal.pntd.0002604PMID: 24392169
24. Schnell MJ, Buonocore L, Whitt MA, Rose JK. The minimal conserved transcription stop-start signal promotes stable expression of a foreign gene in vesicular stomatitis virus. J Virol. 1996; 70: 2318–2323 PMID:8642658
25. Zimmer G. RNA replicons—a new approach for influenza virus immunoprophylaxis. Viruses. 2010; 2: 413–434. doi:10.3390/v2020413PMID:21994644
26. Lichty BD, Power AT, Stojdl DF, Bell JC. Vesicular stomatitis virus: re-inventing the bullet. Trends Mol Med. 2004; 10: 210–216. doi:10.1016/j.molmed.2004.03.003PMID:15121047
27. Halbherr SJ, Brostoff T, Tippenhauer M, Locher S, Berger Rentsch M, Zimmer G. Vaccination with recombinant RNA replicon particles protects chickens from H5N1 highly pathogenic avian influenza virus. PloS One. 2013; 8: e66059. doi:10.1371/journal.pone.0066059PMID:23762463
28. Cobleigh MA, Buonocore L, Uprichard SL, Rose JK, Robek MD. A vesicular stomatitis virus-based hep-atitis B virus vaccine vector provides protection against challenge in a single dose. J Virol. 2010; 84: 7513–7522. doi:10.1128/JVI.00200-10PMID:20504927
29. Klas SD, Lavine CL, Whitt MA, Miller MA. IL-12-assisted immunization against Listeria monocytogenes using replication-restricted VSV-based vectors. Vaccine. 2006; 24: 1451–1461. doi:10.1016/j.vaccine. 2005.05.046PMID:16310294
30. Rose NF, Marx PA, Luckay A, Nixon DF, Moretto WJ, Donahoe SM, et al. An Effective AIDS Vaccine Based on Live Attenuated Vesicular Stomatitis Virus Recombinants. Cell. 2001; 106: 539–549. doi:10. 1016/S0092-8674(01)00482-2PMID:11551502
31. Publicover J, Ramsburg E, Rose JK. A single-cycle vaccine vector based on vesicular stomatitis virus can induce immune responses comparable to those generated by a replication-competent vector. J Virol. 2005; 79: 13231–13238. doi:10.1128/JVI.79.21.13231–13238.2005PMID:16227246
32. Diaz D, Döbeli H, Yeboah-Manu D, Mensah-Quainoo E, Friedlein A, Soder N, et al. Use of the immuno-dominant 18-kiloDalton small heat shock protein as a serological marker for exposure to Mycobacte-rium ulcerans. Clin Vaccine Immunol CVI. 2006; 13: 1314–1321. doi:10.1128/CVI.00254-06PMID: 17021247
33. Marques MA, Chitale S, Brennan PJ, Pessolani MC. Mapping and identification of the major cell wall-associated components of Mycobacterium leprae. Infect Immun. 1998; 66: 2625–2631. PMID: 9596726
34. Vettiger A, Scherr N, Ruf M-T, Röltgen K, Pluschke G. Localization of Mycobacterial Antigens by Immu-nofluorescence Staining of Agarose Embedded Cells. J Mycobact Dis. 2014; 4: 150.
35. Dreyer A, Röltgen K, Dangy JP, Ruf MT, Scherr N, Bolz M, et al. Identification of the Mycobacterium ulcerans Protein MUL_3720 as a Promising Target for the Development of a Diagnostic Test for Buruli Ulcer. PLoS Negl Trop Dis. 2015; 9: e0003477. doi:10.1371/journal.pntd.0003477PMID:25668636 36. Hanika A, Larisch B, Steinmann E, Schwegmann-Wessels C, Herrler G, Zimmer G. Use of influenza C
virus glycoprotein HEF for generation of vesicular stomatitis virus pseudotypes. J Gen Virol. 2005; 86: 1455–1465. doi:10.1099/vir.0.80788–0PMID:15831958
37. Kalhoro NH, Veits J, Rautenschlein S, Zimmer G. A recombinant vesicular stomatitis virus replicon vac-cine protects chickens from highly pathogenic avian influenza virus (H7N1). Vacvac-cine. 2009; 27: 1174–
1183. doi:10.1016/j.vaccine.2008.12.019PMID:19135116
38. Berger Rentsch M, Zimmer G. A vesicular stomatitis virus replicon-based bioassay for the rapid and sensitive determination of multi-species type I interferon. PloS One. 2011; 6: e25858. doi:10.1371/ journal.pone.0025858PMID:21998709
39. Sutter G, Ohlmann M, Erfle V. Non-replicating vaccinia vector efficiently expresses bacteriophage T7 RNA polymerase. FEBS Lett. 1995; 371: 9–12. PMID:7664891
40. Baneyx F. Recombinant protein expression in Escherichia coli. Curr Opin Biotechnol. 1999; 10: 411–
41. Bratschi MW, Bolz M, Grize L, Kerber S, Minyem JC, Um Boock A, et al. Primary cultivation: factors affecting contamination and Mycobacterium ulcerans growth after long turnover time of clinical speci-mens. BMC Infect Dis. 2014; 14: 636. doi:10.1186/s12879-014-0636-7PMID:25433390
42. Lavender CJ, Fyfe JAM. Direct detection of Mycobacterium ulcerans in clinical specimens and environ-mental samples. Methods Mol Biol Clifton NJ. 2013; 943: 201–216. doi:10.1007/978-1-60327-353-4_ 13
43. Fyfe JAM, Lavender CJ, Johnson PDR, Globan M, Sievers A, Azuolas J, et al. Development and appli-cation of two multiplex real-time PCR assays for the detection of Mycobacterium ulcerans in clinical and environmental samples. Appl Environ Microbiol. 2007; 73: 4733–4740. doi:10.1128/AEM.02971-06 PMID:17526786
44. Portaels F, Organization WH. Laboratory diagnosis of buruli ulcer: a manual for health care providers / edited by Françoise Portaels. Diagnostic de l’ulcère de Buruli au laboratoire : manuel destiné au per-sonnel de santé / edité par Françoise Portaels. 2014; Available:http://apps.who.int//iris/handle/10665/ 111738
45. Zimmer G, Locher S, Berger Rentsch M, Halbherr SJ. Pseudotyping of vesicular stomatitis virus with the envelope glycoproteins of highly pathogenic avian influenza viruses. J Gen Virol. 2014; 95: 1634–
1639. doi:10.1099/vir.0.065201–0PMID:24814925
46. Liao W, Lin J-X, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity. 2013; 38: 13–25. doi:10.1016/j.immuni.2013.01.004PMID:23352221 47. Farber DL, Yudanin NA, Restifo NP. Human memory T cells: generation, compartmentalization and
homeostasis. Nat Rev Immunol. 2014; 14: 24–35. doi:10.1038/nri3567PMID:24336101 48. Revill WDL, Morrow RH, Pike MC, Ateng J. A CONTROLLED TRIAL OF THE TREATMENT OF
MYCOBACTERIUM ULCERANS INFECTION WITH CLOFAZIMINE. The Lancet. 1973; 302: 873–
877. doi:10.1016/S0140-6736(73)92005-9
49. Gordon CL, Buntine JA, Hayman JA, Lavender CJ, Fyfe JA, Hosking P, et al. Spontaneous Clearance of Mycobacterium ulcerans in a Case of Buruli Ulcer. PLoS Negl Trop Dis. 2011; 5. doi:10.1371/ journal.pntd.0001290
50. Röltgen K, Bratschi MW, Ross A, Aboagye SY, Ampah KA, Bolz M, et al. Late onset of the serological response against the 18 kDa small heat shock protein of Mycobacterium ulcerans in children. PLoS Negl Trop Dis. 2014; 8: e2904. doi:10.1371/journal.pntd.0002904PMID:24853088
51. Coutanceau E, Legras P, Marsollier L, Reysset G, Cole ST, Demangel C. Immunogenicity of Mycobac-terium ulcerans Hsp65 and protective efficacy of a MycobacMycobac-terium leprae Hsp65-based DNA vaccine against Buruli ulcer. Microbes Infect Inst Pasteur. 2006; 8: 2075–2081. doi:10.1016/j.micinf.2006.03. 009
52. Johnson JE, McNeil LK, Megati S, Witko SE, Roopchand VS, Obregon JH, et al. Non-propagating, recombinant vesicular stomatitis virus vectors encoding respiratory syncytial virus proteins generate potent humoral and cellular immunity against RSV and are protective in mice. Immunol Lett. 2013; 150: 134–144. doi:10.1016/j.imlet.2012.12.005PMID:23261719
53. Martins TG, Trigo G, Fraga AG, Gama JB, Longatto-Filho A, Saraiva M, et al. Corticosteroid-induced immunosuppression ultimately does not compromise the efficacy of antibiotherapy in murine Mycobac-terium ulcerans infection. PLoS Negl Trop Dis. 2012; 6: e1925. doi:10.1371/journal.pntd.0001925 PMID:23209864