WANDA
ROCHA GRONEFELD BAUMANIN
DOENÇA
DAS
CÉLULAS
DE LANG
ERHANS
HISTIOCITOSE X
WANDA ROCHA GRONEFELD BAUMANN
DOENÇA DAS CÉLULAS
DE LANGERHANS
HISTIOCITOSE X
Trabalho de conclusão apresentado ao Curso de
Especialização em Radiologia Odontológica e
lmaginologia da Universidade Federal de Santa
Catarina, como parte dos requisitos obrigatórios
para obtenção do titulo de Especialista em
Radiologia Odontológica e lmaginologia.
Orientador: Prof. Dr. DeImo Tavares
Florianópolis
DOENÇA DAS CÉLULAS DE LANGERHANS
HISTIOCITOSE X
Este trabalho de conclusão foi julgado adequado para obtenção do titulo de Especialista em Radiologia Odontológica e Imaginologia e aprovado em sua forma final pelo Curso de Especialização em Radiologia Odontológica e Imaginologia.
Florianópolis, de de 2007
BANCA EXAMINADORA
Prof. Dr. Delmo Tavares Orientador
Prof. Dr. Membro
Na
busca
do saber
A todos que contribuíram e participaram de uma forma ou de outra para a realização desta etapa do meu aprendizado e que ela seja o inicio de um novo projeto de vida.
Aos meus filhos, Victor Andreas e Marco Antonio, que compartilharam cada momento difícil e souberam compreender o que eu estava buscando e aceitaram as minhas muitas ausências.
A minha irmã Maria Ligia que nos momentos mais difíceis de sua vida não pude fazer-lhe companhia.
A
Berenice, amiga, quiçá nora, que possamos desfrutar de vários momentos juntas no seio familiar.Aos meus funcionários Monica, Terezinha, Roseane e Cristina obrigada pelo carinho e colaboração.
Aos meus pacientes que durante a minha ausência foram realmente pacientes. Aos funcionários do curso de especialização o meu muito obrigada pelo carinho e compreensão.
Aos amigos que sempre me fortaleceram nos momentos difíceis
E a você, mamãe, que do alto de seus 90 anos ainda tem a garra de querer aprender e a vontade de viver dedico-lhe este novo projeto de vida.
A José Simão, meu pai muito amado, que me ensinou o caminho da retidão, hombridade e amor ao proximo, a Luiz Carlos, saudoso irmão, que sempre me orientou na área da saúde, mostrando-me o verdadeiro sentido da missão de HIPOCRATES, meu eterno amor, e a Helaine Mara, minha eterna amizade. (in memonan)
Preferi a sabedoria aos cedros e tronos e, em comparação com ela julguei sem valor a riqueza; a ela não igualei nenhuma pedra preciosa, pois, a seu lado, todo ouro do mundo é um punhado de areia e, diante dela, a prata sera como lama.
Amei-a mais que a saúde e a beleza, e quis possui-la.
Mais que a luz, pois o esplendor que dela irradia não se apaga.
Todos os bens me vieram com ela, pois uma riqueza
incalculável esta em suas mãos.
VOCÊ
se fez presente em
todos os
momentos
firmes e
trimulos.
E, passo a passo, pude
sentir
a sua
mão na
minha
mão,
Transmitindo-me a
segurança necessária
para
enfrentar rneu
carninho
e
seguir..
A
Sua
presença
é qualquer coisa como
a luz
e
a vida,
e
sinto
que
em
meu gesto
existe o
Seu gesto
estei
disposto a perder
o poder
para
fazer
emergir
o
saber
Mestre,
par
opção
ou natureza,
é difícil
de se ter....
Mestre
que,
às
vezes não
TIOScompreende nem
se
faz
compreender...
Mas
existe
aquele que
se interessa, se
responsabiliza, trabalha
com
afinco; que cresce e acrescenta;
que
inquieta e não só
comparece; que caminha
junto
conosco, não
nos
abandonando.
Da busca se
faz
encontrar
Da alegria se
faz amizade
Do saber
encontrei
a
amizade
Da
amizade
encontrei o
saber
Do
Mestre
Maior encontrei o
amor
Dos
mestres
do saber
encontrei
o
caminho
e
a
luz
do
saber
Ao
Prof. Dr. DeImo Tavareso
meu multo
obrigadaMesmo que as dúvidas persistam,
Que a
emoçãonos traia e a
esperançafalte,
Mesmo que as palavras fujam.
Não importa, porque ainda hi tempo...
Para trocar
o silênciopelo sorriso,
Os punhos fechados pelos braços abertos,
Dispensar a distância
eacolher a cumplicidade,
Pedir perdão
esentir saudade...
De tudo e de todos.
Porque agora tudo faz falta...
O
abraço que negamos,
As risadas que não nos permitimos.
Mas ainda há tempo...
do
que
alinhar-se
aos pobres
de
espírito
que não
se
alegram e nem
sofrem
muito, porque vivem em
um
crepúsculo
que não conhece
vitória
ou
derrota."
Imaginologia,
Universidade Federal de Santa Catarina,
Florianópolis.RESUMO
A
Doençadas
Célulasde Langerhans
(DCL) éuma desordem de etiologia incerta
que se apresenta com um comportamento
clinicobastante variado. Seu
aparecimento
advém de
histiócitos quefazem parte do Sistema
Reticuloendotelial,
um dos mais importantes mecanismos de defesa do
organismo. As
doençasque
compõea
DCL são: oGranuloma
Eosinófilo(GE);
Doença
de Hand-Schuller-Christian
(DHSC); e Doençade
Letterer-Siwe (DLS),que fazem parte da
reticuloendoteliose não lipidica. 0levantamento da
situaçãoatual dos conhecimentos da
DCLno meio cientifico baseou-se na
revisãoda
literatura médica
e odontológica.Todos os aspectos como etiologia, incidência,
características
clinicas
e radiográficas, diagnósticodiferencial, tratamento
e prognósticoforam abordados de forma a fazer deste trabalho um manual para
que
o clinicoda Odontologia tenha um eficiente recurso
literáriopara suas
pesquisas nos relacionamentos com estas
doenças.Imaginologia,
Universidade Federal de Santa Catarina,
Florianópolis.ABSTRACT
The Langerhans' Cell's Disease (LCD) is an uncertain
ethiologicdisorder which
shows great variability of clinical behavior. It's happening comes of
histiocytisisfrom the
reticuloendothelialsystem, one of the most important organism's self
defense mechanism. Other diseases contained in the LCD are: the
EosinophilicGranuloma
(EG);Hand-Schuller-Christian's Disease
(HSCD);and
Letterer-Siwe'sDisease (LSD); which are part of the
Nonlipidic retiuloendotheliosis.The study of
LCD's current knowledge situation in the
cientificgroup is sustained by Medical
and
OdontologicalLiterature revision. All aspects, as
ethiology,incidence, clinical
and
radiograficcharacteristics,
diferentialdiagnostic, treatment and prognostic
were studied in such way to make this work a manual so the dentist will have an
efficient literary resource for your researches taken with LC Disease.
Figura 1 — Processo de maturação do monócito 23
Figura 2 - Monócito sanguíneo (mede lOmm - 15mm de diâmetro) 24
Figura 3 - Macrófago em tecido 24
Figura 4 - Células da linhagem dendritica 29
Figura 5- A origem celular dos macrófagos, células de Langerhans, e células dendriticas
das CD34+ células progenitora 32
Figura 6- Célula de Langerhans — Visão por microscópio eletrônico (Observar os grânulos
de Birbeck) 33
Figura 9— Doane! Periodontal 44
Figura 10 - Sindrome de Papillon-Lefèvre 45
Figura 11 - Cisto periapical 45
Figura 12 - Cisto residual 46
Figura 13- Osteomielite crônica supurada 47
Figura 14- Linfoma de Burkitt 48
Figura 15- Fibroma ossificante 48
Figura 16- Mieloma múltiplo 49
Figura 17- Displasia óssea fibrosa 50
Figura 18— Metástases tumorais 50
Figura 19 - Metástases tumorais. 51
Figura 20- Granuloma eosin6filo 51
Figura 23— Aspectos radiográficos do GE. Levantamento periapical 87
Figura 24 - Aspectos radiográficos do GE. Radiografia panorâmica (mesmo paciente da
FIG. 23) 88
Figura 25 - Aspectos Radiográficos do GE. Radiografia em norma lateral — não apresenta
lesão Utica na calota craniana (mesmo paciente da FIG. 23) 88
Figura 26 - Aspectos Radiográficos da DHSL. Radiografia em norma frontal — apresenta
lesão lítica na calota craniana 89
Figura 27 - Aspectos Radiográficos da DHSL. Radiografia em norma lateral — apresenta
lesão litica na calota craniana (mesmo paciente da FIG. 26) 89
Figura 28 - Aspectos Radiográficos da DHSL. Radiografia panorâmical — apresenta perda
óssea alveolar generalizada (mesmo paciente da FIG. 26) 90
Figura 29- Aspectos Radiográficos da DHSL. Radiografia periapical — região de molares
deciduos (D e E) — apresenta perda óssea alveolar severa (mesmo paciente da FIG. 26) 90
Figura 30— Aspectos Clínicos da DHSC. Radiografia panorâmica tirada após 27 anos do diagnóstico inicial da doença de Hand-Schaller-Christian. Nenhuma indicação de lesões
na mandíbula é evidente. Lesões foram tratadas com radiação. 91
Figura 31 — Aspectos Clínicos da DLS. Lesões de pele cobrem o corpo inteiro de um garoto de 10 meses de idade. A cabeça foi enfaixada para absorver a continua
transpiração na cabeça. 91
Figura 32— Aspectos Clínicos da DLS. Visão intra-oral mostra erupção precoce da
Grifico 1 — Distribuiçâo das lesões orals nos c:asos de GE ... 41
Gráfico 2— Distribuição dos sintomas em casos de GE...-.__._. . ... ..._. .. . . ... . 42
Gráfico 3- Proporção das áreas esqueléticas envolvidas da DCL... ... 57
Gráfico 4— Distribuição das lesões orais nos casos de DHSC ... .... ... 59
Gráfico 5 — Distribuição dos sintomas em casos de 59 Gráfico 6— Distribuição das lesões orais nos casos de DLS ....,... .. . . ... 67
Greif:* 7— Distribuição dos sintomas em casos de DLS..,... ... 67
Gráfico 8 - Distribuição de casos de Doença da Célula de Langerhans . ... 75
CM
Célula
Apresentadora de
Antígeno
DCL
Doença
das
Células
de Langerhans
DHSC
Doença
de
Hand-Schüller-Christian
DLS
Doença
de
Letterer-Slwe
GE
Granuloma
Eosinófilo
HC!.
Histiocitose
das
Células
de Langerhans
SFF
Sistema
Fagodtário
Mononuclear
SNC
Sistema
Nervoso
Central
1 INTRODUÇÃO 20
2 PROPOSIÇÃO 21
3 REVISÃO DA LITERATURA 22
3.1 SISTEMA FAGOCITÁRIO MONONUCLEAR 22
3.1.1 Origem do macrófago 23
3.1.2 A origem da histocitose e da desordem histiocistica 31
3.2 HISTORIAMENTO DA DOENga das Células de Langerhans 33
3.2.1 Granuloma eosinofilico 38
3.2.1.1 Conceito 38
3.2.1.2 Etiologia 39
3.2.1.3 Características clinicas e radiográficas 39
3.2.1.4 Diagnóstico diferencial 43
32.1.4.1 Doença periodontal 44
3.2.1.4.2 Síndrome de Papillon-Lefèvre 45
3.2.1.4.3 Cisto periapical 45
3.2.1.4.4 Cisto residual 46
3.2.1.4.5 Cisto primordial 46
3.2.1.4.6 Osteomielite crônica supurada 47
3.2.1.4.7 Linfoma de Burkitt 47
3.2.1.4.11 Metástases tumorais 50
3.2.1.4.12 Granuloma eosinófilo 51
3.2.1.5 Diagnóstico definitivo 52
3.2.1.6 Tratamento 53
3.2.1.7 Prognóstico 54
3.2.2 Doença de Hand-Schuller-Christian 55
3.2.2.1 Etiologia 55
3.2.2.2 Características clinicas e radiográficas 56
3.2.2.3 Diagnóstico clinico 60
3.2.2.4 Diagnóstico diferencial 60
3.2.2.5 Diagnóstico definitivo 61
3.2.2.6 Tratamento 61
3.2.2.7 Prognóstico 61
3.2.3 Doença de Lefterer-Siwe 63
3.2.3.1 Características clinicas e radiográficas 64
3.2.3.2 Tratamento 68
3.2.3.3 Prognóstico 68
4 CONSIDERAÇÕES GERAIS 69
5 CONSIDERAçÕES FINAIS 74
BIBLIOGRAFIA 79
APÊNDICES
APÊNDICE A - DOCUMENTAÇÃO COMPLEMENTAR SOBRE OS ASPECTOS
1 INTRODUÇÃO
Em 1940 um grupo de patologistas americanos reuniu um conjunto de doenças que apresentavam um substrato comum de reação de macráfagos que seriam as retículo-endotelioses, proposição que foi aceita sem julgamento adequado por toda uma geração de hematologistas americanos (OLIVEIRA, 1988).
Somente em 1953, o termo Histicitose X, proposto por Lichtenstein, passou a designar este mesmo conjunto de doenças que compartilhavam as mesmas características clinicas e patológicas.
Entretanto, há uma série de dúvidas a respeito da utilização desta terminologia para descrever diferentes tipos de patologias que representam fases diversas de uma entidade única.
0 conceito que o Granuloma Eosinófilo, Doença de Letterer-Siwe e Doença de Hand-Schüler-Christiam representam formas transicionais evolutivas de uma só entidade patológica tem recebido inúmeras objeções nos últimos anos.
2 PROPOSIÇÃO
3 REVISÃO
DA
LITERATURA
1
Para proporcionar um
melhorentendimento sobre o desenvolvimento da
doença das Células de Langerhans, se faz
necessárioum pequeno
detalhamentosobre o
histiócito,que é o principal componente desta doença.
3.1
SISTEMA FAGOCITARIO
MONONUCLEAR
Sistema Mononuclear
Fagocitário (SMF),Sistema Mononuclear
Macrofágico (SMM)ou Sistema Retículo
-endotelial (SRE) sãotermos coletivos para denominar
um sistema de
distribuiçãode células com grande
expressão fagocitáriaque
atuam sob uma vasta gama de microorganismos. Possuem origens e
fung6escomuns, mas com
localizaçãoem
váriostecidos conjuntivos dispersos pelo
organismo.
Segundo Leeson e Leeson
(apud LAITANO, 1988),o termo Sistema
FagocitárioMononuclear
(SFM)é o mais apropriado, pois, as células que o
comp6e nãoformam
endotélioverdadeiro e
muitasdelas
nãotêm
relaçãocom
Ativação
F
M-edulo Osseo>r-
monob..,-„ r• - lác'toTecidos
•ac..õ(oqo trivodo-
^/ocófcgc
M;crógiios ;SNC) Diferenciação Cé yips de Ki.pie•
.figodo) Mocrarogos ulveolate pulnu5c) Osteocicsk» (3SSOS
Songue
Th
um
retículo;
no entanto, na literatura pertinente sobre Imunologia, adenominação
SistemaRetículo
-endotelial (SRE) ainda tem sido adotada.3.1.1
Origem domacr6fago
O SFM
é
formado por células originárias da linhagem mielóide produzidas pela medula óssea. A forma mais embrionária que compõe esse sistemaé
denominada de célula-tronco ou monoblasto, que através deinúmeras
maturações origina umacoleção
de células da série monocitica. Trowbridgee
Emling (1996) apontaram as células do SMM pelo comportamento macrofágico.
Os monócitos (FIG. 1) são células relativamente imaturas que mantêm a capacidade de se reproduzir, podem permanecer por longo
período
na circulaçãosanguínea
(TROWBRIDGE; EMLING, 1996)e
terão vários destinos. Os fagócitos mononucleares desenvolvem-se na medula óssea, circulam no sangue como monácitose
são encontrados em todos os tecidos do corpo. São células que podem diferenciar-se em formas especializadas em tecidos particulares, comoo
Sistema Nervoso Central (SNC). Possuem um núcleo em forma de ferradura, geralmente com grânulos azurofilicos pálidose
citoplasma contendo lisossomas, vacUolos fagociticos (TROWBRIDGE; EMLING, 1996).(Fonte: HOFLING, 2006. Adaptada de Abbas)
Segundo as condições fisiológicas ou patológicas do organismo, estas células adquirem função fagocitária, transformando-se em fagócito ativo, ou seja, macráfago (FIG. 2 e 3).
(Fonte: HOFLING, 2006. Adaptado de Parham.)
Figura 2 - Monácito sanguíneo (mede lOmm - 15mm de diâmetro)
(Fonte: HOFLING, 2006, Adaptado de Parham.)
Figura 3 - Macráfago em tecido
fundem-se a fim de formar células gigantes e, segundo Brasileiro Filho (2004),
são típicas das inflamações crônicas granulomatosas.
Esses agregados celulares organizados na forma de granulomas podem
conter monáfagos agrupados de diferentes formas:
a)
Células Epitelióides:
quando se agrupam de modo semelhante a
células epiteliais. Nessa situação, esses macrófagos não fagocitam,
embora mantenham a capacidade de participar (englobar partículas
liquidas). Formam-se por estímulos imunogênicos, como por
exemplo, na presença do ovo do
Schistossoma mansoni,
Mycobacterium tuberculosis, Paracoccidioides brasiliensis.
b)
Células Gigantes Multinucleadas:
resultam da fusão de macrófagos.
Há dois tipos:
(i)
tipo Langhans, encontradas de forma característica
na tuberculose, cujos núcleos estão organizados na periferia, e (ii)
tipo corpo estranho, quando os núcleos estão distribuídos
irregularmente no citoplasma, e são comuns em granulomas
induzidos por corpos estranhos imunologicamente inertes.
Segundo Bogliolo (1981) e Trowbridge e Emling (1996), quando o
monócito penetra no tecido conjuntivo ocorre a sua diferenciação em macrófago
recebendo diferentes denominações dependendo do tecido em que se encontrar
(QUADRO 1). No tecido conjuntivo, os macrófagos são também conhecidos como
histiócitos.
CÉLULAS LOCALIZAÇÃO Células precursoras ou Promonócitos Medula óssea
Monoblastos Medula óssea
Pró-monácitos Medula óssea
Monócitos Medula óssea e Sangue periférico
Macráfagos Tecidos:
- Conjuntivo (histiócitos)
- Pulmão (macrófagos alveolares) - Fígado (células de Kupffer)
- Bago (macrófagos livres e fixos)
- Linfonodos (macráfagos livres e fixos) - Medula óssea (macráfagos)
- Cavidades Serosas (macrófagos pleurais e peritoneais)
- Sistema nervoso (micróglia) - Ósseo (osteoclastos)
- Fusão de macrófagos (células gigantes, corpos estranhos)
- Pele (células de Langerhans)
(Fonte: BOGLIOLO, 1981)
Quadro 1 — Componentes do Sistema Fagocitario Mononuclear
Cada uma destas células são macrófagos, mas em determinadas situações revelam características particulares e juntas formam o Sistema Fagocitario Mononuclear (SFM). Conforme citado na Leitura complementar 39, Pato Arte Geral (2001) 2 , as células deste sistema mostram as seguintes características:
a) alta capacidade de fagocitose de células do organismo e de outros corpos;
b) tendências a aderir em superfície;
c) possuem receptores específicos para imunoglobulinas do IgG, participam ativamente das reações de hipersensibilidade, aumentando os mecanismos de ativação, reconhecimento e fagocitose; e
d) alto poder de digestão pela presença de abundantes lisossomos e de
produção de enzimas lisossomiais.
0 Sistema Fagocitário Mononuclear, segundo Haling (2006), possui duas
funções principais, realizadas por dois tipos celulares distintos, ambos derivados
da medula óssea. 0 primeiro conhecido como macrófago fagocitico "profissional",
tem função predominante de remover antígenos particulados, e o segundo, como
Célula Apresentadora de Antigeno (CAA), tem função de internalizar o antígeno e
apresenta-lo as células T. Além de serem importantes células efetoras tanto da
imunidade inata quanto da imunidade adquirida. Braier; Sackmann; Muriel
(1995) incluíram, como CAA, três subgrupos celulares: reticulares dendriticas,
reticulares interdigitantes e de Langerhans.
Pela função de apresentação do antígeno, Unanue e Benacerraf (1986) e
Trowbridge e Emling (1996) caracterizaram os fagócitos mononucleares como
células acessórias nas fases de reconhecimento e ativação dos linfácitos.
Classificam-se como células acessórias os monácitos, macr6fagos e células que
possuem processos dendriticos - como as de Langerhans da epiderme e as
dendriticas encontradas no tecido linfóide.
Para 1-16fling (2006), as principais funções das células acessórias são exibir
o antígeno em uma forma que possa ser reconhecido pelos linfócitos T e produzir
proteínas secretadas e de membrana que sirvam de sinais de ativação das
células "T".
O macráfago ativado tem um grande número de propriedades que lhe proporcionam uma capacidade melhor em matar e digerir os microorganismos fagocitados, em degradar substâncias especiais e em participar das reações imunes. Para englobar microorganismos, os nnacrófagos são ativados por substâncias como INFT, GM-CSF, IL-2 e TNF (TROWBRIDGE; EMLING, 1996). Os mesmos autores apresentaram o resultado da ativação quando comparados a macrófagos não ativados: aumento do tamanho da célula; aumento do número de lisossomas e nas enzimas lisossomais; aumento no número de vilosidades da membrana plasmática; aumento na formação de pseudópodos; quimiotaxia aumentada; maior capacidade de aderência; maior capacidade de fagocitar microorganismos e determinadas substâncias; aumento do metabolismo da glicose; maior produção de metabólitos tóxicos a partir da conversão do oxigênio, aumentando a capacidade de matar os microorganismos ingeridos, como também a capacidade de apresentar antígenos aos linfácitos.
Os macráfagos ativados são também capazes de liberar substâncias que avisam as células mesenquimais que elas devem se acumular e/ou proliferar no local de lesão. Essas substâncias incluem fatores de crescimento derivados de plaquetas e fibronectina, resultando no recrutamento e na proliferação dos fibroblastos e de novos vasos sanguíneos - angiogênese (TROWBRIDGE; EMLING, 1996).
Unanue e Benacerraf (1986) resumiram a função do macráfago em uma combinação de três propriedades: remoção do excesso de antígeno; apresentação de antígeno; e secreção de moléculas estimuladoras.
Segundo Lins et al. (2003), as células dendriticas são capazes de interagir com linfácitos Te Be também modular suas respostas.
A morfologia das células dendriticas (FIG. 4) varia de acordo com o seu estágio de maturação. As células dendriticas maduras caracterizam-se por uma forma irregular, estrelada, exibindo numerosos prolongamentos citoplasmáticos longos e delgados que partem do corpo celular seguindo direções variadas. Já as células dendriticas imaturas apresentam-se com formas arredondadas e prolongamentos citoplasmáticos curtos (LINS et al., 2003).
(Fonte: HbFLING, 2006. Adaptado de Parham)
Figura 4 - Células da linhagem dendritica.
Portanto, as células de Langerhans são células
dendriticas,derivadas da
medula
ósseae
situadas na
regiãosupra-basal da maioria dos
epitélios escamososestratificados. Elas devem agir como células denunciantes do
antígeno
durante a
induçãodas respostas
imunológicas.Embora possuam
funções similares a outras células
dendriticase
macráfagos,as
células
de
Langerhans
sãoespecializadas
e
capazes de migrar até aos
linfonodos,amadurecendo
e
se tornando eficiente
e
na
apresentaçãode
antígenos
aos
linfócitos T
(LOMBARDI et al.,
1993; HOFLING, 2006).Lombardi et al.
(1993)sugeriram que as células de Langerhans
sãocruciais na
induçãodas respostas
imunopatológicasque ocorrem em
níveis
cutâneos,mucosos, ou ambos. Estas células podem representar a primeira linha
de
sensibilizaçãodo sistema
imunológico,levando a um esclarecimento sobre
o
antígeno
ou
fenômeno patológico;entretanto, segundo os mesmos autores,
nãoé
conhecido
o
que leva a
proliferaçãodessas células nas
lesões histiocitárias.De acordo com Lins et al.
(2003),as células de Langerhans apresentam
um baixo potencial de
proliferaçãocelular no interior do
epitélio,e
sendo a
migração
de células progenitoras oriundas da medula
ósseaa
responsávelpelo
aumento do
númerode células de Langerhans no tecido,
e
não um processo de
renovação
celular.
Segundo
Braiere
SackmannMuriel
(1995),um grupo heterogêneo de
alterações
do Sistema
FagocitárioMononuclear
são denominadas Histiocitoses.Cada
síndrome
histiociticaé
resultado de uma
proliferação reativaou
neoplásicade um dos três tipos celulares: reticulares
dendriticas,reticulares
interdigitantesEm 1987, a Sociedade de Histiocitose recomendou dividir as Síndromes Histiociticas em três classes (BRAIER; SACKMANN MURIEL, 1995):
a) classe
I-
Histiocitose de células de Langerhans (apresentador deantigeno)
e
Reticulohistiocitose congênita autoinvolutiva de Hashimoto-Pritzker;b) classe II
-
Histiocitose de fagócitos mononucleares (processador deantígeno);
C)
classe
III-
Histiocitoses Malignas.3.1.2
A origem da
histocitose
e da desordem
histiocistica
\\?
LANGERHANS CELL
DENDRITIC CELL
Sc
MONOCYTE
1111
PHOMONOCYTE ,----+
CFU-GM
MACROffiAGE
„
.
•
•
• • • 411/
. •
Fonte: CLINE, 1994.
Figura 5 - A origem celular dos macr6fagos, células de Langerhans, e células dendriticas das CD34+ células progenitora
Segundo Difranco (1984) os critérios ultra-estruturais para a caracterização das células de Langerhans são os seguintes: núcleo dentado ou lobulado, aparelho de Golgi bem desenvolvido, ausência de melanossomas ou pré-melanossomas, desmossomas e tonofilamentos e presença de grânulos de Birbeck e por serem estes uma característica exclusiva das células de Langerhans, as destinguem dos ceratinácitos e melanácitos, conforme observado
(Fonte NEVILLE et al , 2004, p 494)
Figura 6 - Célula de Langerhans — Visão por microscópio eletrônico (Observar os grânulos de Birbeck)
3.2 HISTORIAMENTO DA DOENÇA DAS CÉLULAS DE LANGERHANS
0 século XIX foi pródigo em descobertas cientificas, principalmente no ramo da Medicina, que revolucionou a maneira de considerar as doenças e lançou as bases da pesquisa médica cientifica, proporcionando um diagnóstico adequado às várias patologias. Dentre os avanços científicos deste século destaca-se a descoberta dos raios X.
Tudo começou em 8 de novembro de 1895, na cidade de Wurzburg, na Bavária, Alemanha, quando William Conrad Roentgen descobriu os raios catódicos que permitiam a visualização da parte interna do corpo humano.
sem danificá-lo, viabilizando uma análise dos dados morfológicos e anatômicos de órgãos e estruturas que, até então, eram exploradas pelas características externas durante a avaliação dos pacientes.
Primeiramente com a tecnologia dos raios X e, posteriormente, com outros meios de exames por imagens, foi possibilitado um diagnóstico mais precoce e preciso de certas doenças que, pelo tardar da identificação, eram tidas como incuráveis (SOARES, apud GRECCO, 2006).
Dentre as doenças que tiveram avanços no seu conhecimento e
diagnóstico através do desenvolvimento da semiotécnica da imagem, se destaca a Doença das Células de Langerhans que é o alvo de interesse no presente estudo.
A enfermidade Doença das Células de Langerhans (DCL) vem despertando a atenção dos pesquisadores por várias décadas. De acordo com os estudos de Hargreaves (1994), a DCL acometeu um personagem ilustre por volta de 1483, o príncipe de Gales Edward V, que faleceu aos 12 anos. Os exames realizados em sua mandíbula mostraram que as lesões eram compatíveis com a DCL, mais precisamente com a do Granuloma Eosinófilo.
Paul Langerhans, em 1868, descreveu uma população de células dendriticas derivadas da medula óssea e situadas na camada suprabasal do epitélio escamoso estratificado, tanto da epiderme como no epitélio da mucosa oral, que exibiam aspectos ultra-estruturais peculiares. Atuam como células
apresentadoras de antígeno (CAA) durante a indução de respostas ao sistema
imunológico e estima-se que sua proliferação anormal leve 6 ocorrência de doenças (LOMBARDI et al., 1993).
1977). Já em 1921,
o
próprio Hand observou outros casos semelhantese
revisou relatosclínicos
de Kay (1906), Christian (1920)e
Schüller (1916), constatando que adoença
descrita era uma nova patologiae
não uma tuberculose, conforme os autores sugeriram (TOMMASI, 1977).As diferentes nomenclaturas descritas por Hand, Schüller, Siwe
e
Letterer geraram uma confusão nomenclatural, que foi, parcialmente, resolvida por Lichtenstein em 1953. Lichtenstein reconheceu que a patologia, pluralmente relatada, consistia em componentes de umadoença
envolvendoo
histiócitoe
que, em cada uma delas, apresentava um padrão
característico.
Ele então propôs a unificação dos termos para Histiocitose X, onde a letra X caracterizavao
fator desconhecido (NEVES et al., 2004).Lichtenstein foi
o
primeiro a introduziro
termo Histiocitose X, em 1953, para designar um grupo de desordens raras relacionadas como
sistemaretículo
endotelial, ou seja, distúrbios proliferativos dos histiócitos ou macráfagos (YU, 1995).
Alguns destes distúrbios, como os linfomas histi6citosos, são claramente malignos enquanto outros, como as proliferações reativas de histiócitos nos linfonodos são benignas (OLIVEIRA, 1988).
Histicitose X
é um
distúrbiodo sistema
reticulo endotelial,caracterizado
por
proliferaçãode
macrófagode aspecto normal, com ou sem
reação inflamatóriaassociada de
eosinófilo, neutrófiloe células
multinucleadas,envolvendo
órgãos,ossos e pele (CARNEIRO FILHO; LEITE; ANDRADE NETO,
2002).
A
etiopatogêniada
DCLnão
estádevidamente esclarecida, embora a
doença possa representar uma reposta
reativaa um defeito na célula mediadora
de imunidade (PRINGLE et al.,
1992).Para alguns pesquisadores a sua origem
pode ser em detrimento das
reaçõesde hipersensibilidade
imunológica,má-absorção intestinal, disfunção
pituitáriae auto-imune
(TOMMASI, 1977).Outros
como
Regezie
Sciubba (2000)e Robbins
(1994)salientaram que a sua etiologia
possa ser de origem
inflamatória.Gabriel
liet al.
(1996)em sua pesquisa sugeriram que a sua etiologia
possa ser de origem infecciosa, possivelmente viral, e trauma local recente
seriam fatores
predisponentespara o desenvolvimento da
DCL.Todavia, atualmente, a doença é mais aceita como sendo um processo
reativo
do que uma desordem verdadeiramente
neoplásica.Alguns autores
discutiram o papel das
citocinasna
patogêneseda doença, e detectaram a
presença dos
linfácitos Tno interior da
lesãoque podem atrair
neutrófllos, eosinófilos, macrófagose
linfócitos CD34da
circulaçãosanguínea e são capazes
de se diferenciarem em células de Langerhans. Recentes
investigaçõesmostram
expressão
anormal de moléculas de
adesãocelular das células da
DCL,o que
contribui numa
migraçãoanormal das células de Langerhans (SOUZA et al.,
1997).
doença
com as células de Langerhans presente na epiderme
emucosa
(ROCHENBACH; CHERUBINI; VEECK, 2004).
0
termo
Histiocitose X écorrespondente a um grupo de patologias que
possuem uma
característicaclinica diferente, mas, com
omesmo
padrão histológico:como no Granuloma
Eosinófilo,forma
crônicalocalizada; na
Doençade
Hand-Schiiller-Christian,forma
crônicadisseminada;
ena
Doençade
Letterer-Siwe,
forma aguda disseminada. Visto que, as células envolvidas nestas
enfermidades apresentam
omesmo
fenótipodas células de Langerhans, a
designação
de
Histiocitosede
Célulasde Langerhans
(HCL)tem sido mais
recentemente utilizada
(TOMMASI, 1977;ROBBINS,
1994;SOUZA et al.,
1997;BARROS; BARROS,
2000;NEVES et al.,
2004).Segundo
Dehner (1989)a
classificaçãoda
HCLinclui as categorias, de
acordo com a Sociedade de
HistiocitoseAmericana:
Histiocitosede Células de
Langerhans
(HistiocitoseX, Granuloma
Eosinófilo, Doençade
Hand-Schüller-Christian,
Doençade Letterer
Siwe); Histiocitosede Células
nãoLangerhans
(síndrome hemofagociticas reativas
associadas a
infecções, linfo-histiocitose eritrofagociticafamiliar, sarcoma
histiócito neoplásico, histicitosemaligna,
e leucemia monocitica-monoblásticaaguda).
Para Carneiro
Filho; Leite;Andrade Neto
(2002)a
Histiocitose X éum
distúrbio
do sistema
reticuloendotelial,caracterizado pela
proliferaçãode
macrófagos
de aspecto normal, com ou sem
reação inflamatóriaassociada de
eosinófilos, neutrófilos e
células
mononuclearesenvolvendo a pele, ossos
e víscerasse
classificandoem Granuloma
Eosinófilo,Síndrome
de
Hand-Schuller-Christian
eSíndrome de
Letterer-Siwe.0
Granuloma
Eosinóffloinclui
lesões ósseas solitáriasou
múltiplas nãoimplicação cutânea,
visceral e da medula
óssea(WALDRON,
1998),sendo esta, a
que melhor representa um processo
neoplásicomaligno (NEVES et al.,
2004).As
observações literáriasexpostas demonstram as dificuldades
históricas,que se prolongam até o presente momento, referentes à definição da etiologia,
classificação,
aspecto
histológicoe nomenclatura da Doença das Células de
Langerhans
(DCL).3.2.1 Granuloma eosinofilico
3.2.1.1
Conceito
É
uma
lesão não neoplásica,benigna agressiva caracteriza-se por uma
proliferaçãointensa de elementos retículo
-histiocitárioscom quantidade
variávelde
linfácitos, eosinófilos, neutrófilos,células
plasmáticase células gigantes
nucleadas. 30
nome
eosinofflicose deve à presença de numerosos
eosincifilosdispersos
na
lesão (MANTESSO; RAITZ, 2006).É
a forma mais suave e
freqüenteda
DCLsendo do tipo
crônicolocalizada
apresentando
lesões ósseas únicas (monostóticas)ou
múltiplas (poliostóticas)que acometem crianças e adultos jovens (KELLY,
1962;NEVES et al.,
2004).Segundo Castro
(2000) nãose observa o envolvimento visceral no GE.
Sedano
et al.
(1969)mostraram que as
lesões liticasocorrem nos ossos
do crânio e mandíbula principalmente na
regiãoposterior.
Pereira et al.
(2004) afirmaramque
oGE
solitáriona
infância éconsiderado
raro.Yu et al.
(1995)demonstraram que
oGE afeta
ocrânio,
vértebras,pelve
sendo que os maxilares são os mais afetados.
a entidade da
DCL querepresenta um total maior de incidência de todas
as
reticulo-endotelioses não lipidicas,perfazendo um total de
50%a
60%segundo Hartman
eColonel
(1980).3.2.1.2 Etiologia
Embora haja um consenso entre os pesquisadores sobre a célula de
Langerhans ser a origem da
doença,a sua etiologia, em termos de fatores
desencadeantes,não
estáainda bem esclarecida. Segundo
Menacheset al.
(1998) oGE tem sua etiologia ainda desconhecida.
Para
Galliniet al.
(1988), oGE
éuma variedade benigna do
mieloma múltiploenquanto que outros pesquisadores
oconsideram uma
osteite nãopurulenta
emais precisamente uma
histiomatose inflamatória.Laitano (1988) considerou o
GE como um processo
reativo e neoplásico.3.2.1.3 Características clinicas e radiográficas
Ao se examinar um paciente,
o cirurgiãodentista deve ficar atento
às lesõesorais presentes, para chegar ao
diagnósticodo GE.
0
paciente pode apresentar estado febril,
irritabilidade,perda de peso,
cefaléia, anorexia,
adenopatiaduras
e
móveis,sendo
indiciode que
o
GE
é
poliostótico (LAITANO, 1988).Geralmente
nãoexiste sintomatologia dolorosa
e
quandopresente esta
é
difusa
e
localizada. Leal Filho et al.
(2003)em sua
amostra,citou que
o
GE
apresentou sintomatologia dolorosa em
87%dos casos,
o
que foi corroborado
por Pereira et al.
(2004).Segundo Oliveira
(2004),o
GE pode apresentar dor ou
ser
assintomático,bem como apresentar hipersensibilidade
dentária.Os sintomas bucais
sãoos primeiros fatores que induzem
o
paciente a
procurar
o
tratamento
odontológico (TOMMASI, 2002).0
primeiro sinal
é
a mobilidade dental sem causa aparente,
o
que se
recomenda exame
anátomo-patológicodos tecidos moles
curetadosjunto aos
dentes
quandodo tratamento periodontal ou
exodontia,visando um correto
diagnóstico;
assim como exame
radiográficocompleto do esqueleto
e
cintilografia ósseacom Tc-99 bem como
tomografia computorizadasegundo
Franjul
et al.
(1998), jáque
o
exame
radiográfico nãonos
dáum quadro preciso
do GE devido à sua
semelhança
com outras
doenças
radiolkidas.Hartman
e
Colonel
(1980)efetuaram interessante trabalho com
114pacientes portadores de
DCLe
encontraram
89casos de GE em que as
lesõesbucais se apresentavam como uma massa
palpávele
dolorosa.
0
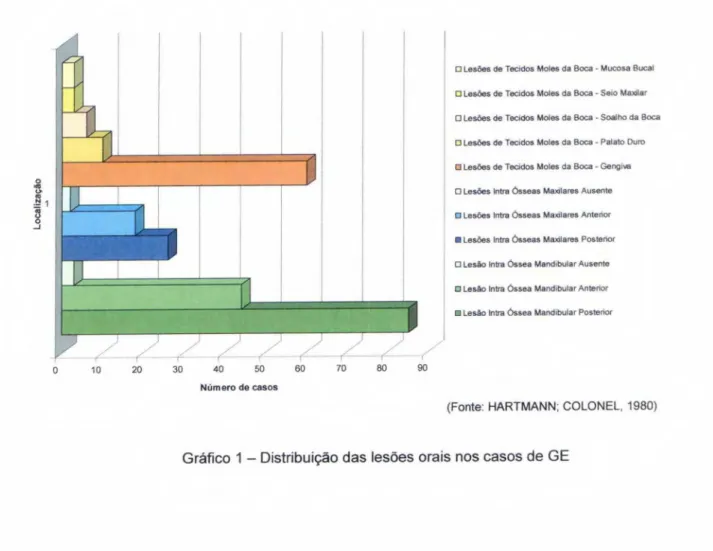
GRAF.
1mostra a
distribuiçãode
lesõesencontradas no referido
trabalho, onde predominaram as
lesões intra-ósseasmandibulares em região
posterior, seguidas de
lesõesno tecido mole-gengiva.
0 númeromédio de
lesõesr
10 20 30 40 50
Número de casos
70
ID Lesões de Tecidos Moles da Boca - Mucosa Bucal
o Lesões de Tecidos Moles da Boca - Selo Maxilar
O Lesões de Tecidos Moles da Boca - Soalho da Boca ID Lesões de Tecidos Moles da Boca - Palato Duro
Cl Lesões de Tecidos Moles da Boca - Genghe
o Lesões !we Osseas Maxilares Ausente
O Lesões Intra Osseas Mar-dares Antenor
• Lesões Intra Osseas Mar-tiaras Posterior
o Lesão Intra Ossea Mandibular Ausente
O Lesão Intra õssea Mandibular Antenor
Lesão Intra Ossea Mandibular Postenor
(Fonte: HARTMANN: COLONEL, 1980)
Gráfico 1 — Distribuição das lesões orais nos casos de GE
Ocorre retardamento de cura após exodontia (OLIVEIRA, 2004).
As manifestações bucais são úlceras necróticas, edema, halitose,
mobilidade dental com perda ou esfoliação precoce do elemento dental
(TOMMASI, 1977; TOLEDO et al., 1997).
0 GRAF. 2 mostra a distribuição dos sintomas encontrados nos casos
O Parestesia
• Hemorragia Gengival
O Infecção de Vincent
El Dificuldade na Mastigação
0 Ma Cicatrização
• Alteração do Hato e Gosto
O Dor ou Sensibilidade - Ulceração Oral
0 Dor ou Sensibilidade - Dentes Perdidos
• Dor ou Sensibilidade - Gengivite
OAumento de Volume Gengival
o 5 10 15 20 25 30 35 40 45 50 Número de Casos
(Fonte: HARTMANN: COLONEL, 1980)
Gráfico 2 — Distribuição dos sintomas em casos de GE
Existe uma predominância pelo gênero masculino na proporção de 2:1 segundo Pereira et al. (2004) e Leal Filho et al. (2003). Para Mesa Zarate et al. (1983) esta proporção também é de 2:1, apesar de estudos anteriores como o de Camargo et al. (1988), que não especifica o índice do GE sobre o gênero.
Radiograficamente a destruição do osso alveolar é um dos sinais mais característicos do GE podendo simular uma periodontite localizada severa ou uma infecção periapical (RAPP; MOTTA, 2000).
Segundo Higashi; Shiba; Ikuta (1991) as imagens radiográficas dos ossos alveolares apresentam uma aparência de "comida de mariposas" com resultante aparência de dentes flutuantes, soltos no alvéolo.
A imagem radiográfica
é
radiolúcida arredondada com margens bem menos definidas que um cistoe
os dentes aparecem como se estivessem flutuando no ar (ISHII et al., 1992; SOUZA et al., 2000).Os ossos gnáticos são os mais envolvidos, principalmente a
mandíbula,
provocando uma área mais localizada de destruição óssea periapical na região posterior (SEDANO et al., 1969; GIONA et al., 1997; PASLER; VISSER, 2005), sendo que autores como SIGALA et al. (1972) não determinaram a sua localização.
Segundo Pasler
e
Visser (2001), as lesões osteolíticas são pouco definidase
se confluem; na mandíbula, reconhece-se uma linha basal delimitante, sombreada, em forma de guirlandae
muitas vezes com reações osteoblásticas, envolvimento da lamina dura que estádestruída
mostrando os dentes flutuantes,o
que leva as fraturas patológicas.Tommasi (1977) descreveu uma reação periosteal junto a áreas osteolitica,
e
que a expansão ósseaé
rara.Shafer; Hine; Levy (1987) declararam que em desdentado ocorre uma área de radiolucidez uniforme, sem osteogênese reacional,
o
que lembra uma neoplasia.3.2.1.4 Diagnóstico diferencial
0 GE tem seu diagnóstico diferencial com as seguintes patologias ósseas, segundo Ishii et al. (1992): cistos,
doença
peridontalavançada,
osteomelites, linfoma de Burkitt, displasia fibrosa óssea, mieloma múltiploe
metástasesZachariades et al. (1987) apresentaram como diagnóstico diferencial do GE outras entidades patológicas tais como: doença periodontal, síndrome Papillon-Lefèvre, cistos odontogênicos, querubismo, osteomielites, mieloma múltiplo, ameloblastoma, mixoma odontogênico, hemangioma, defeito ósseo traumático, depresão da glândula sublingual, linfoma, sarcoma osteogênico, fibroma ossificante, displasia fibrosa, osteogênese imperfeita e metástase óssea.
3.2.1.4.1 Doença periodontal
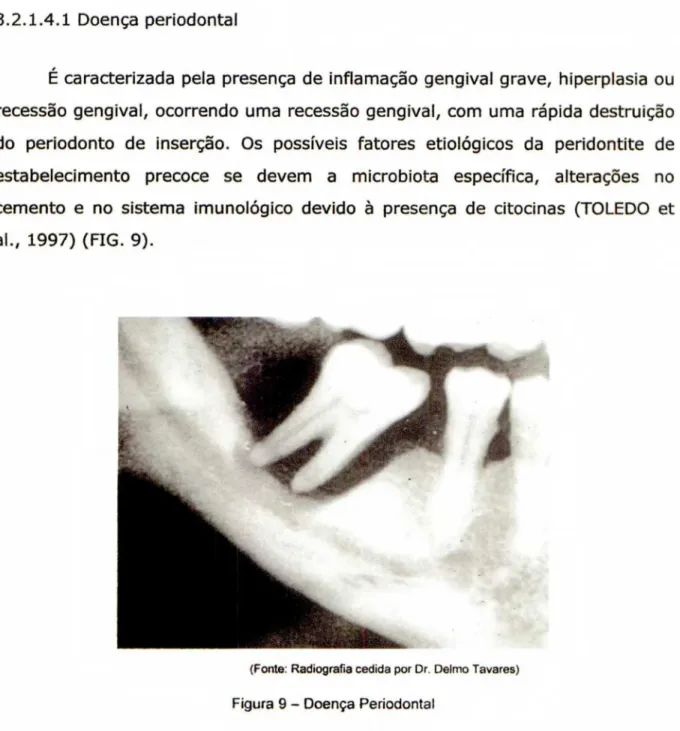
É caracterizada pela presença de inflamação gengival grave, hiperplasia ou recessão gengival, ocorrendo uma recessão gengival, com uma rápida destruição do periodonto de inserção. Os possíveis fatores etiológicos da peridontite de estabelecimento precoce se devem a microbiota especifica, alterações no cemento e no sistema imunológico devido â presença de citocinas (TOLEDO et al., 1997) (FIG. 9).
(Fonte: Radiografia cedida por Dr. DeImo Tavares)
(FonteI TOLEDO et al , 1997)
3.2.1.4.2
Síndrome
de Papillon-LefèvreOcorre uma progressão rápida
e
inflamação gengival severa. É uma doençahereditária rara com
característica
autossiimica (TOLEDO et al., 1997) (FIG. 10).Figura 10 - Síndrome de Papillon-Lefevre
3.2.1.4.3 Cisto periapical
A imagem
é
radiolkida com osteogênese racional, bem circunscrita naregião periapical do elemento dental (TOMMASI, 1977) (FIG. 11).
(Fonte: LANGLAND: LANGLAIS, 2002, p.416)
3.2.1.4.4 Cisto residual
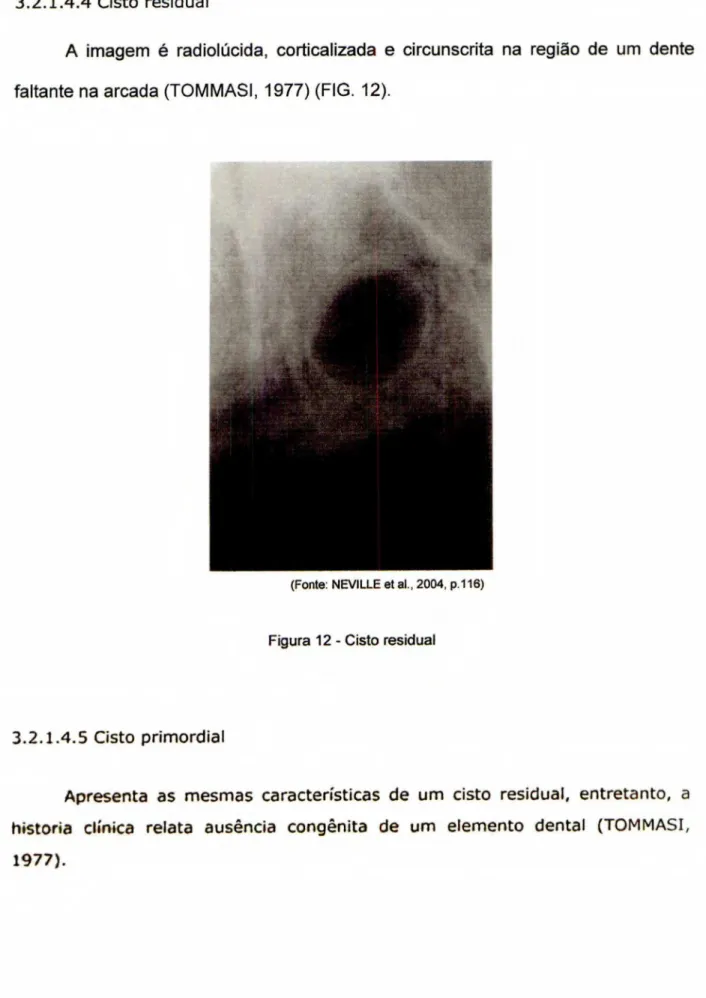
A imagem é radiolúcida, corticalizada e circunscrita na região de um dente
faltante na arcada (TOMMASI, 1977) (FIG. 12).
(Fonte NEVILLE et al 2004, p116)
Figura 12 - Cisto residual
3.2.1.4.5 Cisto primordial
Apresenta as mesmas características de um cisto residual, entretanto, a historia clinica relata ausência congênita de um elemento dental (TOMMASI,
3.2.1.4.6 Osteomielite crônica supurada
Radiograficamente as áreas radiolúcida são extensas, com limites imprecisos, aparecem áreas com seqüestros ósseos (necróticos), sem expansão óssea (TOMMASI, 1977) (FIG. 13).
(Fonte: PASLER et al., 2001, p.234)
Figura 13 - Osteomielite crônica supurada
3.2.1.4.7 Linfoma de Burkitt
(Fonte: NEVILLE et al., 2004)
(Fonte: NEVILLE et al., 2004, p.502)
Figura 14 - Linfoma de Burkitt
3.2.1.4.8 Fibroma ossificante
Na fase inicial apresenta área radiolkida, sem cápsula, com limites moderadamente definidos e tem predileção pelo sexo feminino (NEVILLE et al.,
2004).
3.2.1.4.9 Mieloma múltiplo
Apresenta áreas radiolúcidas pequenas e arredondadas, sem
corticalização, afeta mais de um osso, apresenta sintomatologia dolorosa, incidindo em pessoas acima de 40 anos e sua confirmação se faz através de exame sanguíneo em que a proteína Bence-Jones está presente (NEVILLE et al.,
2004) (FIG. 16).
(Fonte: NEVILLE et aI., 2004, p.504)
Figura 16 - Mieloma múltiplo
3.2.1.4.10 Displasia óssea fibrosa
Na sua fase inicial apresenta área radiolúcida com limites imprecisos
Figura 17 - Displasia óssea fibrosa
3.2.1.4.11 Metástases tumorais
Devido à semelhança de imagem radiolúcida deve-se averiguar se há ou houve história de lesões malignas em outras áreas (FIG. 18 e 19).
(Fonte: Radiografia cedida pelo Dr. DeImo Tavares).
(Fonte: PASLER et al., 2001, p.295)
Figura 19 - Metástases tumorais.
3.2.1.4.12 Granuloma eosinófilo
A FIG 20 apresenta granuloma eosincifilo de acordo com Tommasi (1977).
3.2.1.5 Diagnóstico
definitivo
Os aspectos histológicos do GE são considerados os mais clássicos devido ao acúmulo focal de eosinófilos (TOMMASI, 1997).
Segundo Regezi e Sciubba (2000) existe uma proliferação de células grandes com núcleos ovais e riniformes, cromatina dispersa com abundante citoplasma eosinófilo nos corte histológicos corados com hematoxilina-eosina.
No estudo imuno-histoquimico observaram que as células anormais foram positivas para HLA-DR, proteína 5-100, CD1 e principalmente ocorria a presença de grânulos de Birbeck quando vistos sob a luz de microscopia eletrônica (REGEZI; SCIUBBA, 2000).
Nos exames de cintilografia óssea observa-se uma alta concentração de radiofármacos nas áreas envolvidas (SOUZA et al. 1997).
Estas identificações confirmaram estudos anteriores de que o GE apresenta um fenótipo característico e sua origem a partir das células de Langerhans (BARROS; BARROS, 2000).
Laitano (1988) cita Leccisotti; Licci; Mirisola (1983) que apresenta uma classificação histológica de Engelbert Holm que nos dá uma melhor idéia da evolução do GE. A classificação se baseia em fases:
a) fase
proliferativa
- hiperplasia com eosinofilia e proliferaçãodifusa-histiocitária;
b) fase
granulomatosa -
presençade células gigantes, numerosos
eosinófilos e macráfagos
em
ação fagocitária, oque
dáum aspecto
C)
fase
xantomatosa -
presença de células xantomatosas, que nada mais
são que histiócitos em atividade lipofágica;
d)
fase fibrosa
-
corresponde à evolução final, onde ocorre a proliferação
de fibroblastos com sucessiva esclerose local.
3.2.1.6 Tratamento
A forma de tratamento recomendada pela maioria dos pesquisadores varia
desde a não interferência pela sua regressão espontânea até cirurgias aliadas
radioterapia, quimioterapia.
Zegarelli et al. (1981), e confirmado por Regezi e Sciubba (2002) são os
que preconizaram a regressão espontânea do GE.
A forma mais indicada para o tratamento do GE é a remoção cirúrgica feita
através de curetagem ou ressecção (KESSLER et al., 2001).
As lesões individuais podem ser controladas efetivamente com curetagem
ou baixa doses de radiação segundo Regezi e Sciubba (2002) e Ishii et al.
(1992).
Quando
o
tratamento
cirúrgico nãofor
possível,mas existir um
envolvimento
poliostótico,a radioterapia
é
a escolhida, sendo que a dose de
radiação
deve ser escolhida de acordo com a idade do paciente
e o
local afetado
variando de
3a
10 Gy(BAER;
BENJAMIM, 1975;HARTMAN; COLONEL,
1980;KESSLER et al.,
2001).A quimioterapia tem sido recomendada
quandose observa um
envolvimento sistêmico ou
quandoocorre uma
lesão secundáriade uma
lesãolocalizada.
Utilizam-se agentes
citotásticos, antimetabólicose
imunossupressoresentre os quais os mais citados
sãoa
Vinblastina, Vincristina, Ciclofosfamida, Metotrexatoe
Predinizolona (MENACHESet al.,
1998;KESSLER et al.,
2001; REGEZIe
SCIUBBA, 2000; VISCAYAet al.,
2005).3.2.1.7 Prognóstico
Todos os autores pesquisados
são unânimesquanto ao
prognóstico
do GE
que
é
de bom a excelente desde que ocorra um
diagnósticocorreto para evitar
tratamentos inadequados (CHASE et al.,
1974).indicado um acompanhamento por muito tempo para afastar a
possibilidade de recidiva (CHASE et al.,
1974; RAPIDISI, 1978;ISHII et al.,
1992).
Para Hartman
e
Colonel
(1980)pode ocorrer recidiva,
e
segundo
Sedanoet al.
(1969)pode existir a possibilidade de evoluir para a
doença
disseminada
3.2.2
Doença de Hand-Schuller-Christian
A
doença
de Hand-Schuller-Christian
(DHSC)se apresenta como a forma
crônica
da
DCLtambém denominada Granuloma
Eosinófilico multifocal.Apresenta uma tríade
clássicarepresentada por
lesões osteoliticasnos
ossos
membranosos, exoftalmiae
diabetes
insipidus
(TOMMASI, 1977).A diabetes
insipidus
provém do
infiltradode
linfócitosna
hipófiseque leva
também
à ocorrência de nanismo,
infantilismoe
polkiria,e
a
exoftalmiaé
proveniente do infiltrado de
histiócitosna
órbita(BAER; BENJAMIN,
1975;BARROS; BARROS,
2000).Podem ocorrer
alteraçõesem outros tecidos como
o fígado, vísceras,
pele,
nódulos linfáticos, pulmões
e
mucosa (BAER; BENJAMIN,
1975).Apresenta uma maior
incidênciaem
criançase
jovens adolescentes
(HARTMAN; COLONEL,
1980).Além
da
tríade
clássica,os pacientes apresentam estado febril,
erupções descamativasdifusas semelhante à seborréia, sobretudo no couro cabeludo
(TOMMASI, 1977);
nos canais auditivos ocorrem
otitese
mastoidites;também
ocorrem
infecções respiratóriase
fibroses pulmonares
(ARAÚJO
JR; SOUZA,
2004).
3.2.2.1
Etiologia
27% (137)
11% (56)
4% (19) <1% (1) 8%(42)
8%(41)
6%(32)
<1%(2)
3%
(1
5)<1%(1)
<15(0(1) 3.2.2.2 Características clinicas e radiograficas
As manifestações clinicas surgem antes dos 5 anos de idade e em adulto jovem, apresentado envolvimento visceral, porém, não tão difusa e disseminada como a forma disseminada aguda.
Envolvem todo o esqueleto afetando o crânio, mandíbula, pelves, costelas, tibia, escápula, fêmur e úmero, além de poder apresentar lesões cutâneas (TOMMASI, 1977).
Kilpatrick et al. (1995), a partir do estudo de 263 casos de DCL, mapearam a distribuição das lesões ósseas em todo o esqueleto, o que pode ser observado na FIG. 21 e seu correspondente no GRAF. 3.
(Fonte: KILPATRICK et al . 1995)
27%
•
Cega•
Trunco o Mentirre inferiores D Men-toms Superiores14%
(Fonte: KILPATRICK et al., 1995)
Gráfico 3 — Proporção das areas esqueléticas envolvidas da DCL
A mandíbula apresenta grande destruição óssea, ocorrendo mobilidade dental e perdas precoces dos dentes ou exposição radiculares bem como hálito fétido e salivação abundante (CHASE et al., 1974).
Os sintomas mais característicos são a poliúria, polidipsia, perda de peso e dermatite seborrêica (ARAÚJO JR; SOUZA, 2004).
Segundo Zachariades et al. (1987), os sinais e sintomas da DHSC compreendem estomatite generalizada, hemorragia gengival, úlceras e necrose da mucosa oral, destruição óssea progressiva do processo alveolar, perda prematura dos dentes e assimetria facial.
Cranin e Rockman (1981) relataram que a presença dos diabetes insipidus