UNIVERSIDADE DE LISBOA
FACULDADE DE CIÊNCIAS
DEPARTAMENTO DE BIOLOGIA ANIMAL
Role of lgr family members in AER renewal during
limb bud development
Diana Sofia Chapela Duarte Pires
Mestrado em Biologia Evolutiva e do Desenvolvimento
2011
UNIVERSIDADE DE LISBOA
FACULDADE DE CIÊNCIAS
DEPARTAMENTO DE BIOLOGIA ANIMAL
Role of lgr family members in AER renewal during
limb bud development
Diana Sofia Chapela Duarte Pires
Dissertação orientada por: Professor Doutor Joaquín Rodríguez-León
Professor Doutor Élio Sucena
Mestrado em Biologia Evolutiva e do Desenvolvimento
2011
I
Acknowledgments
Não poderia deixar de agradecer a todos aqueles que contribuiram para que esta etapa da minha vida fosse alcançada.
Em primeiro lugar, um muito obrigado ao Joaquín! Obrigada pela oportunidade desta experiência, pelos conhecimentos transmitidos, pela disponibilidade, pelo optimismo com que acreditaste em mim e pelo incentivo, pela alegria e humor contagiante. Boss, you rock \m/ ! Ao Professor Élio que aceitou ser meu orientador interno e se demonstrou disponível para qualquer dúvida ou problema.
Ao Instituto Gulbenkian de Ciência, uma instituição com excelentes condições e espírito científico.
À Catarina, por toda a ajuda e apoio que me deu durante ‘os meus primeiros passos’.
À Joana Monteiro, Rita Aires, Rita Félix e ao Arnon por toda a ajuda e companheirismo (viva o turno da noite pessoáu!!). Ao Moisés que se mostrou sempre disponível quando precisei à Andreia, Inês e Aybuke, obrigada por todo o apoio e boa disposição!
À Raquel Tomás, que me ensinou a dar os ‘primeiros passos’. Obrigada pela tua ajuda incansável, pela paciência e apoio. Obrigada por teres sempre acreditado em mim, pelas maluqueiras, pela amizade!
À Rita Lopes, obrigada Ritinha! Pela companhia, pela alegria e boa disposição.
Aos amigos, Susana Santos, Daniel McGuire, Joana Lima e Ana. Ao Bernardo Chaves. Obrigada pela amizade e carinho, pelo apoio incondicional e pela força que me transmitiram. Vocês foram uma peça chave nesta etapa.
E finalmente, à família! Em especial à minha mãe, pela força e coragem que sempre me transmitiste, pelo amor… pelo alicerce que és na minha vida. À grande mulher que foi a minha avó Laura, ao meu avô e ao meu Pai, que ficariam orgulhosos se me pudessem acompanhar nesta concretização pessoal. À minha irmã e ao melhor sobrinho do mundo, Gabriel. Obrigada por fazerem parte da minha vida!
II
Abstract
During limb development, the proximal-distal outgrowth is controlled by a thickening of ectodermal cells at the distal tip of the limb, termed apical ectodermal ridge (AER). This transient embryonic structure is essential for the patterning and limb outgrowth, being a conserved feature in vertebrate development and it is also important to maintain proliferation in adjacent tissues before their differentiation. Despite AER induction and maintenance are orchestrated by complex interactions between the FGF, WNT/β-catenin and BMP signaling pathways, little is known about the molecules involved in the maintenance of the proliferation versus apoptosis and the renewal of its cells during development. In recent studies, it has been showed by our lab that one of these molecules, oct4, could be involved in the control of proliferative balance within the AER cells.
In this thesis we present evidences of the involvement of two more molecules in this process,
lgr5 and lgr6, which are known as adult stem cells markers. In here, we describe the
expression pattern of lgr5 and lgr6 during limb bud development using the chicken embryo as a model and show that their expression patterns in the limb bud are consistent with the areas of cell proliferation within the AER. Moreover, we performed lgr5 gain-of-function experiments through in ovo electroporation and studied the relationship of lgr5 and lgr6 with different signaling pathways known to be involved in the AER induction and maintenance. The phenotypes obtained point to the involvement of lgr5 in the maintenance of a proliferative niche in the AER. We also present here, evidences showing that lgr6 is controlled by WNT signaling. Our results support a model in which lgr5 and lgr6 control the activation and maintenance of a niche of undifferentiated cells that are in continuous proliferation at the base of AER. This niche can be responsible for the renewal of AER until it disappears by massive programmed cell death.
IV
Resumo
O desenvolvimento do membro em vertebrados segue um conjunto de multiplas interacções celulares que são regidas ao longo de três eixos principais: o anterior-posterior (AP), o proximal-distal (PD) e o dorsal-ventral (DV).
O crescimento e diferenciação do primórdio do membro dependem do estabelecimento e manutenção de 3 centros organizadores: a crista ectodérmica apical (AER, do inglês, Apical Ectodermal Ridge), um espessamento da ectoderme morfologicamente distinto que se situa na fronteira dorso-ventral na parte mais distal do esboço do membro (regula o crescimento ao longo do eixo próximo-distal); a zona de actividade polarizante (ZPA do inglês, Zone of Polarizing Activity) no mesênquima, na margem posterior do primórdio, não apresentando características morfológicas particulares (regula o crescimento ao longo do eixo antero-posterior); e a ectoderme que não constitui a AER, que está envolvida na determinação da polaridade dorso-ventral do primórdio do membro (Capdevila & Izpisua Belmonte, 2001; Martin, 2001)
A AER é estrutura embriónica transiente e é essencial para a padronização do crescimento do membro, sendo uma característica conservada no desenvolvimento em vertebrados, importante para manter as células mesenquimais adjacentes em proliferação até que a sua diferenciação ocorra. Assim, a remoção cirúrgica desta crista ectodérmica apical conduz a morte celular no mesênquima e consequentemente truncagem esquelética (Bénazet J. and Zeller R., 2009; Capdevila and Izpisua Belmonte, 2001; Ros M. and Fernandez-Teran M., 2008). A indução, a formação e a expansão das células precursoras da AER, envolvem uma série de sinais provenientes da mesoderme e de um conjunto de moléculas sinalizadoras, nomeadamente factores de crescimento fibroblástico (FGFs, do inglês Fibroblast Growth Factors), WNTs e proteínas morfogenéticas do osso (BMPs, do inglês Bone Morphogenic Proteins). Por sua vez, a sua manutenção requer um equilíbrio entre proliferação e morte celular e uma rede de complexas interacções entre as vias de sinalização FGF, WNT/β-catenin e BMP (Bénazet J. and Zeller R., 2009; Ros M. and Fernandez-Teran M., 2008). No entanto, pouco se sabe acerca das moléculas e dos mecanismos envolvidos neste processo. Recentemente, foi demonstrado no nosso laboratório que uma dessas moléculas envolvidas no controlo do equilíbrio proliferativo dentro das células da AER e na manutenção do nicho de células responsáveis pela sua renovação é o oct4. Até então, oct4 tinha sido descrito como sendo necessário para o estabelecimento e manutenção da população de células pluripotentes para a embriogénese no humano e no ratinho (Campbell P. et al. 2007).
Nesta tese, apresentamos evidências do envolvimento de outras duas moléculas, lgr5 e lgr6, conhecidas como marcadores de células estaminais adultas em diversos epitélios como o do intestino e da pele.
No intestino, as células são geradas de novo nas criptas e perdem-se por apoptose no topo das villi. A homeostasia do epitélio no intestino adulto é coordenada por diversas vias de sinalização tal como Notch, WNT, BMP and Hedgehog. A via de sinalização WNT é uma das mais importantes, desempenhando um papel crucial na manutenção e activação da proliferação dos reservatórios de células estaminais. A sua remoção genética em ratinho bloqueia a renovação epitelial no intestino (Haegebarth A. and Clevers H., 2009; Sato T. et al., 2009; Barker N. et al., 2007).
V
Lgr5 é apenas expresso nas células da base da cripta, constituindo-as assim células estaminais
multipotentes que ainda estão indiferenciadas. Para além disso, através da sua activação pela via de sinalização WNT, está envolvido no aumento massivo da proliferação epitelial, quer no epitélio intestinal quer na pele (Sato T. et al., 2009; Carmon K. et al., 2011). Este gene apresenta ainda uma expressão restricta em outros tecidos como o olho, o cérebro, a glândula mamária, os orgãos reprodutivos e o estômago.
O processo de auto-renovação é geralmente descrito como um evento celular paralelo à proliferação, diferenciação e apoptose. Este processo é controlado por vias genéticas intrínsecas que, por sua vez, são reguladas por sinais que vêm do microambiente envolvente, designado de nicho, no qual as células estaminais residem (Zhang and Li, 2005; Haegebarth A. and Clevers H., 2009). Curiosamente, o sistema pelo qual a homeostasia do epitélio do intestino é regida, apresenta alguma similaridade com dinâmica celular no processo de auto-renovação da AER.
Lgr6 por sua vez, também está descrito como marcador de uma população de células
estaminais da pele, estando envolvido na capacidade proliferativa, contudo, diversos estudos referem que ele parece actuar através de uma via de sinalização não dependente de WNT (Snippert H. et al., 2010, Cédric Blanpain, 2010; Barker N. and Clevers H., 2010).
Neste trabalho, nós descrevemos o padrão de expressão destes dois genes ao longo dos estádios de desenvolvimento do embrião de galinha e mostramos como o seu padrão de expressão no membro, quando a AER está formada, co-localiza com as áreas de proliferação celular. Para além disso, realizaram-se experiências de ganho-de-função de lgr5, por electroporação in ovo e estudou-se a relação deste gene com diferentes vias de sinalização através da utilização de microsferas embebidas em diversas moléculas que se sabe estarem envolvidas na manutenção e indução da AER.
No que concerne à sobrexepressão de lgr5 mostramos evidências de que esta resulta no aumento dos níveis de β-catenina na ectoderme, levando ao aparecimento de projecções da AER pre-formada e de posteriores fenótipos com peças esqueléticas extra e mesoderme ectópica, o que vem reforçar a hipótese de lgr5 estar envolvido na proliferação celular.
Ao contrário do que está descrito por diversos autores (Snippert H. et al., 2010, Cédric Blanpain, 2010; Barker N. and Clevers H., 2010), que referem que lgr6 parece não ser controlado pela via de sinalização WNT, os nossos resultados apresentam ainda evidências claras do contrário. De facto, lgr6 é controlado pela via de sinalização WNT. Neste trabalho, elucidamos ainda as interacções existentes entre este gene e outras vias de sinalização, mostrando que lgr6 é negativamente controlado por BMPs, FGFs e Ácido Retinóico (RA, do inglês Retinoic Acid).
Deste modo, propomos um modelo no qual, lgr5 e lgr6, estão envolvidos na activação e manutenção da proliferação de um nicho de células estaminais na base da crista ectodérmica apical, responsável pela sua renovação.
No entanto, é ainda necessário realizar mais experiências de modo a clarificar o papel destes genes. Deste modo, uma das primeiras experiências seria a de perda de função do lgr5 através da electroporação com RNA de interferência e a sobreexpressão e subregulação de lgr6 no membro.
Palavras chave: Lgr5 e lgr6, desenvolvimento do membro, crista ectodérmica apical, via de
Table of Contents
I – Introduction ... 1
I.1 - Vertebrate limb development... 1
I.2 - Developmental dynamics of the AER ... 2
I.3 - The AER as a model for the study of epithelial renewal ... 4
I.4 - WNT signaling and maintenance of stem cell pluripotency ... 5
I.5 - LGR family ... 5
I.5.1 - R-spondins function as ligands of the orphan receptors LGR4 and LGR5 ... 6
I.5.2 - Lgr5, WNT signaling and stem cells ... 7
I.5.3 - Lgr6 as marker of skin stem cells ... 8
Objectives ... 10
II - Materials and Methods ... 11
II.1 - Animal model ... 11
II.2 - Embryo collection, fixation and storage ... 11
II.3 - Experimental manipulation of the limb ... 12
II.3.1 - Bead implantation ... 12
II.3.2 - In ovo electroporation ... 12
II.4 - Experimental construction of RNA interference (RNAi) ... 13
II.4.1 - RNAi cloning ... 15
II.5 - Cloning of full length lgr5 ... 16
II.5.1 - RNA extraction from tissues... 16
II.5.2 - 1st Strand DNA Synthesis ... 16
II.5.3 -Amplification of the fragment to be cloned ... 16
II.5.4 - Extraction and purification of a band in Agarose gel ... 17
II.5.5 -Ligation ... 17
II.5.6 - Bacterial transformation ... 18
II.5.7 - Transformants analysis ... 18
II.5.8 -Extraction of plasmidic DNA – Minipreps ... 18
II.5.9 - Digestion of the Plasmid ... 18
II.5.10 - Sequencing ... 19
II.5.11 - Amplification of the amount of DNA – Midipreps ... 19
II.6 - In situ hybridization ... 19
II.6.2 - Plasmid Linearization ... 20
II.6.3 - Agarose gel electrophoresis ... 20
II.6.4 - Phenol:Chloroform extraction and RNA/DNA precipitation with ethanol ... 20
II.6.5 - In vitro DIG-labelled anti-sense RNA probe transcription ... 21
II.6.6 - Whole-mount In situ hybridization ... 21
II.7 - Histological analysis ... 22
II.7.1 - Tissue processing and gelatin embedding ... 22
II.8 - Proliferation assays ... 22
II.8.1 - BrdU incorporation and detection ... 22
II.9 - Imaging ... 23
II.10 - Limb morphological analysis ... 23
II.10.1 - Alcian green staining ... 23
III – Results ... 24
III.1 – Lgr5 ... 24
III.1.1 - LGR5 phylogenetic analysis ... 24
III.1.2 - Lgr5 is expressed during limb development ... 25
III.1.3 - Cell dynamics ... 26
III.1.4 - Lgr5 overexpression ... 27
III.1.5 - Lgr5 downregulation ... 28
III.1.6 - Lgr5 regulation ... 28
III.1.6.1 - Regulation of lgr5 by FGF signaling ... 29
III.1.6.2 - Regulation of lgr5 through WNT signaling ... 29
III.1.6.3 - Regulation of lgr5 by BMP signaling ... 30
III.1.6.4 - RA involvement in lgr5 expression ... 30
III.2 - Gallus gallus’s Lgr6 ... 30
III.2.1 - Lgr6 is expressed in the AER ... 30
III.2.2 – Regulation of Lgr6 ... 32
III.2.2.1 - Lgr6 regulation by FGF signaling ... 32
III.2.2.2 - WNT signaling controls lgr6 expression in AER ... 33
III.2.2.3 - BMPs negatively controls lgr6 expression ... 33
III.2.2.4 - RA activity controls lgr6 expression ... 34
IV- Discussion ... 35
IV.1 - Lgr5 and lgr6 in AER development and renewal ... 35
IV.3 - Lgr5 and lgr6: the effect of FGF and WNT signaling modulating their expressions ... 36
IV.4 - Lgr5 is necessary but not sufficient for proper AER formation and maintenance ... 37
IV.5 - Proposed model for lgr5 and lgr6 activity ... 38
Concluding remarks ... 39
References ... 40
Appendix I – Embryonic stages of Gallus gallus ... 44
Appendix II – Buffers, Solutions and Media ... 45
Appendix III – Plasmid maps ... 48
Introduction
1
I – Introduction
The study of the complex processes and signaling networks that govern development from the initial relative simplicity of a fertilized egg could be defined as the main goal of Developmental Biology.
During development, the formation of a new organism results from the coordinated combination of multiple processes including growth, patterning and cellular differentiation through a temporal and spatial regulation of gene expression and cell behavior. However, the processes by which an undifferentiated field of cells acquire spatial pattern and undergo coordinated differentiation are not fully understood (Berge et al., 2008, Ros M. and Fernandez-Teran M., 2008).
To understand these processes, vertebrate limb development can be a powerful model, since it is not a vital organ for embryonic life. For this reason, it is easy to perform genetic or surgical manipulations in this model like the removal or transplantation of tissues and that is compatible with embryo survival.
I.1 - Vertebrate limb development
The initial vertebrate limb bud is formed by a large embryonic field where cells receive proliferative and positional information through signals of instructive signaling centers (organizers) which in turn, are firmly regulated both spatially and temporally.
The developing limb emerges from the flank of the embryo as a bud of undetermined mesenchymal cells covered by surface ectoderm, around stage 12HH. After the initial budding, limb development follows multiple intercellular interactions and the main ones are directed by three signaling centers: the apical ectodermal ridge (AER), a specialized thickened region of the ectoderm at the distal tip of the limb bud; the zone of polarizing activity (ZPA) which corresponds to the posterior mesenchyme of the bud and the non-ridge ectoderm (Bénazet J. and Zeller R., 2009; Capdevila and Izpisua Belmonte, 2001; Ros M. and Fernandez-Teran M., 2008).
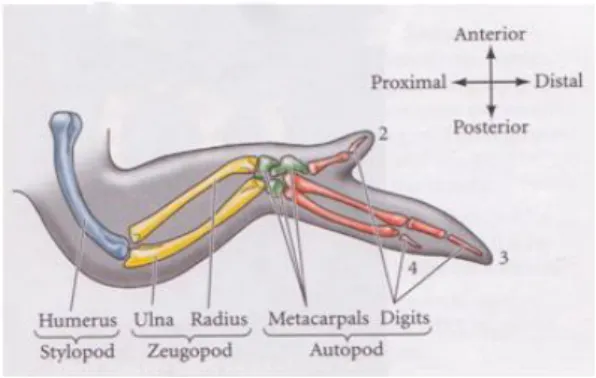
The limb bud in vertebrates is patterned during the embryonic development by these organizer centers along three main axes: the proximal-distal (PD), dorsal-ventral (DV) and anterior-posterior (AP). As a final result, three limb segments are formed: the stylopod that contains the humerus or femur; the zeugopod, distal to the stylopod, containing the radius and ulna or tibia and fibula; and the autopod, at the tip, that contains the carpal/tarsal, metacarpal/metatarsal bones and fingers/toes (Bénazet J. and Zeller R., 2009).
The anterior-posterior (AP) identity is specified by the ZPA through morphogenetic Sonic
hedgehog (Shh) signaling. Additionally, this morphogenetic signaling at the posterior limb is
under control of several genes like dHand, 5’Hox-, Gli3 and fgf8 as well as RA signaling from the flank. Moreover, the Hox gene hoxb8 was also proposed to be required for the initiation of
Shh expression, acting as its upstream regulator. On the other hand, the dorsal-ventral (DV)
polarity is controlled by the activity of the non-AER limb ectoderm where the dorsal ectoderm secretes wnt7a. This molecule is the candidate to specify dorsal fate, controlling the
Introduction
2 expression of lmx1 in the underlying dorsal mesenchyme while in the ventral ectoderm en-1 acts as a ventralizing factor, preventing wnt7a from being expressed in the ventral part of the limb bud (Bénazet J. and Zeller R., 2009; Capdevila and Izpisua Belmonte, 2001).
Figure 1 – Skeletal pattern of the chick wing. The digits are numbered 2, 3 and 4 (from Gilbert, 2006)
The third axis, the proximal-distal axis, is controlled during limb development by the AER which elicits several growth factors and, among others, fibroblast growth factors (namely FGF8, FGF4, FGF9, FGF17). This thickening of ectodermal cells, formed by a pseudostratified epithelium at the distal tip of the limb, is a conserved feature responsible for keeping the underlying mesenchymal cells in a proliferative and undifferentiated state (the so-called progress zone - PZ). Thus, surgical removal of the AER causes a developmental arrest, leading to apoptosis in the mesenchyme and resulting in a skeletal truncation. This truncation affects more proximal segments when the AER is removed at early stages and affects distal elements if removal is performed at later stages (Yu K. and Ornitz D. 2008; Bénazet J. and Zeller R., 2009).
Notwithstanding, although several genetic and cellular mechanisms related with AER induction and maintenance during limb development have been described, little is known about the link between them and the maintenance of its structure. Some studies have shown that a localized region of the epidermis gives rise to AER precursor cells in response to mesenchymal signals. Besides that, other elements like somites, intermediate mesoderm and lateral plate mesoderm are also involved in AER induction. These tissues act as a source of FGFs (fibroblast growth factors) and WNTs that induce AER formation what, in turn, also appears to be strongly associated with the formation of the dorsal-ventral axis ( Satoh et al., 2010; Berge et al., 2008).
I.2 - Developmental dynamics of the AER
The transient structure of the AER undergoes a series of morphogenetic changes during its life span that can be divided into three main phases. A first phase starts with the specification of the AER precursor cells; then, a middle phase, in which the mature AER is well established and, finally, a final phase, where AER flattens and regresses.
Regarding the formation of this structure, two processes have to be considered: AER induction and maturation. The first one is based on the induction of the AER precursor cells in the surface ectoderm that will migrate to the dorsal-ventral boundary to condense and form the
Introduction
3 AER. This process is driven by complex interactions, involving the FGF, WNT/β-catenin and BMP signaling pathways that operate within the ectoderm and between the mesoderm and ectoderm of the prospective limb bud. Several studies have shown that fgf10, a member of the FGF superfamily, is the factor provided by the mesoderm to initiate the process of AER induction (Yu K. and Ornitz D., 2008). Some authors suggest that two members of the WNT family, wnt2b and wnt8c, induce fgf10 mesenchymal expression in a β-catenin-dependent process, which in turn, with the support of bmp4, will prompt the AER establishment by the initiation of fgf8 expression in the AER precursor cells. Supporting this hypothesis, it has been also described that the targeted disruption of fgf10 or its receptor fgfr2b produces embryos in which fgf8 is not detected, indicating that the induction of the AER precursor cells did not take place and, consequently, the AER is not formed (Kawakami Y. et al., 2001; Capdevila and Izpisua Belmonte, 2001; Ros M. and Fernandez-Teran M., 2008). Thus, there must be a reciprocal positive feedback loop that maintains both fgf8 and fgf10 expressions. This way,
fgf10 signals from LPM to the ectoderm to induce another Wnt gene, wnt3a, which in turn,
induces fgf8 expression in the surface ectoderm. In summary, fgf8 from the IM controls fgf10 in the LPM, which induces fgf8 in the overlying surface ectoderm. Therefore, three wnt genes that signal through β-catenin mediate the FGF8/FGF10 loop that leads to limb initiation and AER induction, providing a specific example of cross-talk between signaling pathways (Ros M. and Fernandez-Teran M., 2008; Kawakami Y. et al., 2001, Tomás, A.R., in preparation).
In addition, limb bud initiation is also affected by retinoic acid (RA) whose synthesis and signaling are restricted to the proximal part of the limb (by FGF activity) after limb bud appearance. Thus, AER-FGF morphogenetic activities in the distal mesenchyme seem to be a result of direct RA antagonism in the proximal part of the limb field to repress fgf8 expression. This prevents excessive FGF activity and inhibits limb induction (Mercader et al., 2000; Zhao et
al., 2009). Additionally, studies performing ectopic RA signaling or inactivating CYP26B1 (an
enzyme involved in the degradation of RA) showed that the synthesis of RA occurs in the proximal mesenchyme and spreads to the distal side of the limb bud where it is actively degraded, resulting in a proximal-distal gradient of RA activity (Bénazet J. and Zeller R., 2009). Regarding the second process, maturation of the AER, it results in the formation of the characteristic, thickened structure where pre-AER cells in the ventral ectoderm migrate towards the distal tip and undergo a compaction process at the DV boundary level. Thus, the DV boundary establishment in the ectoderm is also a necessary condition for AER formation. Once DV axis is induced and during limb outgrowth, the dorsal ectoderm express wnt7a which plays an essential role in the control of DV patterning by imposing the expression of lmx1 in the underlying dorsal mesenchyme. In addition, BMP signals from the lateral mesoderm, specify the ventral ectoderm by activation of en-1 expression which prevents wnt7a from being expressed in the dorsal side of the limb bud (Capdevila and Izpisua Belmonte, 2001; Ros M. and Fernandez-Teran M., 2008).
The mature AER is then maintained through a tuned balance between proliferation and cell death for an additional period of 2-3 days, while mesenchymal skeletal progenitors continue to proliferate and differentiate until a fully patterned limb emerges. This equilibrium is genetically controlled but little is known about the molecules involved in this process. After that, the AER regresses via programmed cell death and eventually flattens to a simple cuboidal epithelium
Introduction
4 (Lu, P. et al. 2008; Tomás, A.R., in preparation). This regression is under the control of BMP signaling since overexpression of Noggin or Gremlin, two BMP antagonists, leads to an abnormal AER persistence (Capdevila 1998, Merino et al. 1999, Zuñiga et al. 1999).
I.3 - The AER as a model for the study of epithelial renewal
An important goal in developmental and stem cell biology is to clarify the mechanisms that allow embryonic progenitor cells to choose a specific path for differentiation or to maintain their pluripotency.
Stem cells (SCs) are key pillars in the biology of the tissue throughout life and they are involved in the homeostasis maintenance of tissues that are constantly replacing their cell populations, like skin, blood or intestinal epithelium. Moreover, they form a reservoir of cells that can be activated after tissue injury. Thus, a stem cell pool can be defined by two essential properties: the ability to maintain itself throughout long periods of time (self-renewal) and the potential to generate all differentiated cell types of the pertinent tissue (multipotency) both in vitro and in
vivo (Pardo M. et al., 2010; Haegebarth A. and Clevers H., 2009; Barker N. and Clevers,
H.,2010).
Self-renewal process is generally described as a parallel cellular event of proliferation, differentiation and apoptosis, being controlled by intrinsic genetic pathways that are subject to regulation by extrinsic signals from the microenvironment, called niche, in which SCs reside. Regarding this, several studies have been shown that protein regulatory network that includes the pluripotency factors NANOG, OCT4 and SOX2 are crucial in the maintenance of the undifferentiated and pluripotent state of mouse and human embryonic stem (ES) cells. Besides that, WNT signaling has also been proposed to maintain the self-renewal and pluripotency of this mouse ES cells (Zhang and Li, 2005; Haegebarth A. and Clevers H., 2009).
AER is an epithelial transient structure maintained by a tuned balance between proliferation and cell death and is sustained throughout development by a coordinated network of signaling pathways during limb development. However, little is known about the behavior of AER cells and the dynamic of cell renewal at the most distal tip of the limb during development.
From the molecules described above, oct4 has been shown to be required to establish and maintain the pluripotent cell population necessary for embryogenesis in mouse and human (Campbell P. et al. 2007). Consistent with that, our laboratory have found evidence pointing at
oct4 as a key molecule controlling the proliferative balance within the AER cells and
maintaining a niche responsible for the renewal of AER structure (Tomás A. R. et al.,
manuscript in preparation). However, many of other players have to be involved in this
process. Therefore, AER seems to be an important model for the study of cellular renewal related to the action of organizer centers during development.
Introduction
5
I.4 - WNT signaling and maintenance of stem cell pluripotency
The WNT pathway is one of the main signaling pathways that functions in the early development to regulate body axis specification, germ layer formation and organogenesis. Beyond that, the WNT pathway has also been reported to promote ES cell pluripotency and regulate the self-renewal of ES cells. Related to this, several studies showed strong genetic evidence involving this family in crucial steps during the regulation of epithelial SCs in the intestinal tract as well as in the regulation of SCs in hair follicle bulge. Moreover, in the SCs of the skin, WNT signaling appears to be critical not only for regulating tissue homeostasis but also for wound repair (Sokol, S., 2011; Haegebarth A. and Clevers H., 2009).
According to the accepted scheme of the canonical WNT pathway, WNT proteins act through various Frizzled receptors and low-density lipoprotein co-receptores LRP5/LRP6 to activate Disheveled. In addition, LRP6-mediated activation of the canonical pathway also occurs in response to a new family of ligands termed R-spondins (Bell et al., 2008). Once Disheveled is activated, it inhibits the activity of glycogen synthase kinase-3 (GSK3) enzyme, leading to the inactivation of the β-catenin-degradation complex. This way, stabilized β-catenin associates with transcription factors of the TCF/lymphoid enhancer factor 1 (LEF1) family, and this complex seems to be essential for target gene activation. In the absence of WNT ligands, β-catenin is degraded and maintained at a low cytoplasmic level (Chai R. et al, 2011; Sokol, S., 2011).
WNT ligands and receptors have a proven role in pluripotency, however, despite the major molecular players of WNT pathways being conserved, the mechanisms involved in this signaling pathway with stage-specific and cell context-dependent outcomes remain unclear. Nevertheless, several studies have been shown that WNT signaling plays different, and sometimes opposite, roles at different stages of the same process. (Sokol S., 2011).
One of the models proposed to explain how WNT signaling maintains pluripotency in ES cells consists in the formation of a complex between OCT4 and β-catenin that leads to the activation of OCT4-depend reporters. This binding acts in two manners: in axis formation it antagonizes the WNT pathway and in the ES cells it seems to have a stimulatory role in WNT-dependent ES cell self-renewal. Moreover, it has been described that ES cell pluripotency and self-renewal are promoted when the pathway is upregulated at any level, by adding exogenous WNT3A ligand, inhibiting GSK3 activity, by overexpressing β-catenin or depleting TCF3 (Sokol S.,2011).
I.5 - LGR family
Glycoprotein hormone receptors, including thyroid-stimulating hormone (TSH) receptor, follicle-stimulating hormone (FSH) receptor and leutinizing hormone (LH) receptor, belong to the large G protein-coupled, 7-transmembrane protein superfamily and are unique in having a large N-terminal extracellular ectodomain. This ectodomain contains a series of leucine-rich repeats and, in these receptors, it is crucial for binding to the glycoprotein hormones (Park J. et
Introduction
6 In 1998, Hsu et al. cloned two molecules that are related to these hormone receptors. The two novel G protein-coupled receptors were termed leucine-rich repeat-containing, G-protein-coupled receptors -4 and -5 (LGR4 and LGR5). Moreover, in 2000, the same investigators identified other two members of this family, LGR6 and LGR7 (Barker N. and Clevers H., 2010; Hsu S. et al., 2000).
Recently, phylogenetic analysis confirmed that there are 3 LGR subgroups: the LH, FSH and TSH glycoprotein hormone receptors. The ligands of the first subgroup are defined structurally by the presence of a cysteine domain which is common to a range of extracellular signaling proteins. In the third subgroup (LGR7 and LGR8), ligands belong to a different class, consisting in small heterodimeric peptides with homology to insulin, including the pregnancy hormone relaxin and insulin-like (Barker N. and Clevers H., 2010). However, the ligands for LGR4, LGR5 and LGR6 are still object of study.
The ectodomain of LGR4, LGR5 and LGR6 consist of a central array of multiple leucine-repeats (18 in LGR4 and LGR5, and 13 in LGR6) that are flanked by N-terminal cysteine-rich sequences and the junctions between this ectodomain and the first transmembrane region are highly conserved between these three LGRs. It is predicted that the leucine-rich repeat region of these three LGRs adopt a horseshoe shape that provides a binding site for a peptide ligand. Recent studies give us a clue about this subject, demonstrating that LGR4 and LGR5 bind the R-spondins (RSPOs), a group of secreted proteins with high affinity that enhance WNT/β-catenin signaling (Carmon K. et al.,2011; Lau W. et al.,2011). Moreover, it seems that these three receptors evolved early during evolution since homologous proteins are found in invertebrates, such as sea anemone, mollusk, the nematode Caenorhabditis elegans and
Drosophila melanogaster (Park J. et al., 2005; Barker N. and Clevers H., 2010).
I.5.1 - R-spondins function as ligands of the orphan receptors LGR4 and LGR5
R-spondin family proteins are a novel class of signaling ligands, consisting in a group of four secreted proteins (RSPO1-4) that induce canonical WNT signaling. These proteins contain an N-terminal signal peptide, furin-like cysteine-rich domains, a thrombospondin type 1 repeat, and a C-terminal low-complexity region enriched with positively charged amino acids (Nam J. et al., 2007; Aoki M. et al., 2008; Carmon K. et al., 2011).
RSPO1-4 share 40-60% identity between each other and beyond acting synergistically with extracellular components of the WNT pathway, they also function as a class of ligands independent of WNT proteins, leading to downstream activation of β-catenin-dependent genes. Furthermore, it has been described that RSPO1-4 stimulate the proliferation of intestinal crypt stem cells both in vivo and in vitro through enhancement of WNT/β-catenin signaling via β-catenin stabilization (Nam J. et al., 2007; Carmon K. et al., 2011). Interestingly, it was also described that R-spondin2 is specifically expressed in the AER of mouse developing limb, where appears to be essential for its maintenance (Aoki M. et al., 2008; Bell S. et al., 2008).
Regarding to the third subgroup of LGRs (LGR4, LGR5 and LGR6), its endogenous ligands and signaling mechanisms are not fully understood. However, it was recently discovered that
Introduction
7 RSPOs potentiate WNT/β-catenin signaling by functioning as high-affinity ligands of LGR4 and LGR5. This way, it was proposed a model in which activation of LGR4 and LGR5 by RSPOs leads to an increase in LRP6 co-receptor phosphorylation and consequently enhanced β-catenin activation, but the exact mechanism behind this process remains unclear.
Notwithstanding, it is well established that WNT/β-catenin signaling requires internalization of the WNT co-receptors and the sequestration of glycogen synthase kinase-3 (GSK3) inside multivesicular endossomes. Therefore, it is possible that the RSPO-LGR complex enhances the internalization of the frizzled-WNT-LRP6 signalosome into multivesicular endosomes, leading to enhanced LRP6 phosphorylation (Nam J. et al., 2006; Nam J. et al., 2007; Carmon K. et al., 2011; Lau W., et al., 2011; Bell S. et al., 2008).
I.5.2 - Lgr5, WNT signaling and stem cells
Lgr5 (leucine-rich repeat containing G protein-coupled receptor 5), also known as HG38,
GPR49 and FEX, is a WNT target gene which has been recently identified as a novel stem cell marker of the intestinal epithelium and hair follicle. Furthermore, it also exhibits a high restricted expression in a variety of other tissues such as the eye, brain, mammary gland, reproductive organs and stomach; representing this way a general marker for adult SCs (Haegebarth A. and Clevers H., 2009; Sato T. et al., 2009; Barker N. et al., 2007; Carmon K. et
al., 2011).
In the stem cell niche of hair follicle, lgr5 is expressed in active cycling cells. The hair follicle stem cells seem to be regulated by WNT signaling, which in turn, has been reported to be critical not only for regulating tissue homeostasis but also for wound repair (Haegebarth A. and Clevers H., 2009; Leushacke M. and Barker N., 2011; Barker N. and Clevers Hans, 2010).
In addition, in the intestine, cells are newly generated in the crypts and are lost by apoptosis at the tips of the villi. Lgr5 is exclusively expressed in cycling crypt cells and these lgr5 positive cells constitute a multipotent stem cell niche that generate all cell types of the epithelium (Schepers et al., 2011; Leushacke M. and Barker N., 2011; Sato T. et al., 2009)
As mentioned before, the WNT signaling plays an important role in the maintenance and activation of proliferation of SC reservoirs and the crucial role of this pathway in the intestine,
Introduction
8 was established when its genetic ablation in mouse blocked the epithelial renewal in the small intestine. Additionally, lgr5 seems to act through its ligands (RSPOs) to regulate the canonical WNT pathway which, when stimulated, induces a massive increase in epithelial proliferation (Lau W et al., 2011; Carmon K. et al., 2011; Leushacke M. and Barker N., 2011).
This way, lgr5 is firstly activated through the canonical WNT signaling pathway (Chai R. et al., 2011) and forms a complex with R-spondin ligand (RSPO-LGR5 complex) which, in the target, enhances the internalization of the frizzled-WNT-LRP6 signalosome, leading to enhanced LRP6 phosphorylation and consequently enhanced β-catenin activity (Carmon K. et al., 2011; Bell S.
et al.2008).
Interestingly in the intestine, while WNT/β-catenin signaling leads to an increased cell proliferation; some authors hypothesized that lgr5 plays a negative role in this signaling pathway but is also required for cell proliferation. Thus, lgr5 not only would enhance WNT/ β-catenin signaling but also would accelerate the degradation of pLRP6 and β-β-catenin, suggests that activation of lgr5 in the intestine would play a dual role generating essential signals for cell proliferation and for the down-regulation of WNT pathway (Carmon K. et al., 2011; Leushacke M. and Barker N., 2011).
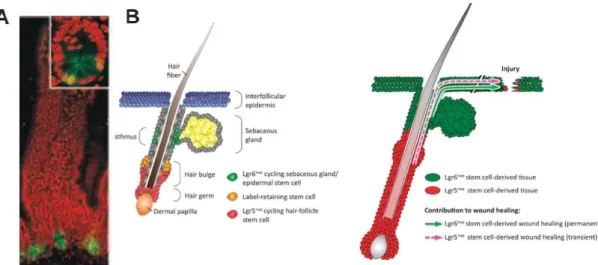
Figure 3- Adult stem cell compartments ensure epithelial homeostasis in the intestine adult hair-follicles and skin and contribute to tissue regeneration following injury. (A) Lgr5 expression is restricted to CBC cells interspersed
between the Paneth cells at the base of the crypt. (B) Lgr5 and lgr6 positive stem cells in the hair follicle and in the skin, respectively (from Barker N. and Clevers H., 2010 and Leushacke M. and Barker N., 2011)
I.5.3 - Lgr6 as marker of skin stem cells
Lgr6 (leucine-rich repeat containing G protein-coupled receptor 6) a close homolog of lgr5, is
known as a marker for a distinct population of stem cells that gives rise to all lineages of the skin. This gene, exhibits a localized expression in the developing hair-pegs of embryonic skin and in a restricted domain in the adult hair follicle, just below the sebaceous gland. However, unlike lgr5, lgr6 does not appear to be controlled by WNT signaling (Snippert H. et al., 2010, Cédric Blanpain, 2010; Barker N. and Clevers H., 2010).
Moreover, several studies have been described that lgr6 positive skin stem cells permanently contribute to newly generated epidermis, during wound repair. These lgr6 positive cells were
Introduction
9 also described to contribute to the hair follicle neogenesis within the wound epithelium which implies plasticity under damage conditions (Leushacke M. and Barker N., 2011).
The discovery of lgr5 and lgr6 as markers in several adult stem cell populations in the mouse has contributed to our understanding of adult stem cell biology in rapidly self-renewing tissues. Importantly, the dynamics described for the epithelium renewal in the intestinal epithelium and at the most distal tip of the limb during development seems to share some similarities. Having these data in our hands, lgr5 (and also its homologous) could be good candidates for understanding the maintenance of a cell niche responsible for the renewal of the AER.
Introduction
10
Objectives
The apical ectodermal ridge (AER) is maintained through a tuned balance between proliferation and cell death and is sustained throughout development, however little is known about the mechanisms and molecules involved in this process.
The cell dynamics described for the epithelium renewal in the intestine (and in others epithelia) and at the most distal tip of the limb during development seem to share some similarities. Since we are interested in understanding the mechanisms behind apical ectodermal ridge renewal during development and we have some candidate genes for this process, namely lgr5 and lgr6, we specifically aim to:
1- Elucidate the role of lgr5 and lgr6 during limb development and AER maintenance. 2- Study the relationship between lgr5 and lgr6 and AER cell renewal.
3- Understand the relationship between the genetic cascades responsible for AER activity and lgr5 and lgr6 expressions.
Materials and Methods
11
II - Materials and Methods
II.1 - Animal model
Avian embryos, specially the chicken (Gallus gallus) embryo, have been important experimental model organisms to perform genetic and developmental studies. This animal model is easy to keep and produce a considerable number of embryos, whose development occurs outside the body of the mother. Moreover, it is also possible to develop chick embryos in culture outside the egg and the access to the embryo during development allows experiments involving surgical manipulation and application of proteins or chemicals that interfere in the process of development.
As vertebrates, they have common developmental processes with that of humans (although they present many differences), like the molecular basis of limb development, whose process is similar both in humans and birds.
II.2 - Embryo collection, fixation and storage
White legorn chicken (Gallus gallus) fertilized eggs were supplied from Sociedade Agrícola da
Quinta da Freiria, S. A.. These eggs were incubated at 37.5˚C - 38˚C with 40% humidity. Chicken
embryos used in this work ranged from stages 11 to stages 35 and were classified according to the Hamburger and Hamilton developmental table (Hamburger and Hamilton, 1951).
Before beginning any procedure and to ensure that embryos were maintained in an RNAse free environment and aseptic conditions, microsurgery instruments were sterilized at 120˚C for two hours. Then, chicken eggs were windowed when embryos reached the proper stage. The embryos were dissected from the yolk/vitelline sac and transferred to a Petri dish containing cold Phosphate buffer saline (PBS). Depending on the stage of embryos, the head and viscera were removed and from stage 26HH onwards only the hindlimbs were collected. After this, the embryos were fixed overnight at 4˚C in 4% Paraformaldehyde (PFA) to preserve embryo structures and to prevent mRNA degradation. On the following day, embryos were washed twice in a PBT solution (1X PBS; 0.1% Tween-20) and were dehydrated through a crescent series of methanol solutions in PBT (25%, 50%, 75% and 100%). Finally, they were stored in absolute methanol at -20˚C (for at least 6 hours prior use) in order to stabilize RNAses, prevent transcript degradation and avoid the formation of water crystals that would damage cellular integrity.
Materials and Methods
12
II.3 - Experimental manipulation of the limb
Embryos are incubated always with their blunt pole up and, this way, the air chamber is placed at the top of the egg making the manipulation process easier. Before performing each experimental manipulation, eggs should be withdrawn from the incubator 1 hour in advance, in order to decrease heart beatings and embryo blood pressure. This way, if any little vessel injury occurs we would have a faster healing process.
Moreover, it should be mentioned that in these all manipulation techniques we performed our experiments in the right limb bud, leaving the left limb as a control.
II.3.1 - Bead implantation
To assess the involvement of lgr5 in AER renewal and its regulation by other molecules, we studied the effects of local application of different molecules through implantation of beads soaked in a particular protein/drug solution.
Beads were implanted at stage 21HH (Hamburger and Hamilton, 1951) chicken embryos at the most distal tip of the limb, subjacent to the AER. For this experiment, a small window was opened in the egg and then we performed a small slit in the vitelline membrane above the embryo with fine forceps, exposing the right limb bud. After this, a little incision was made at the distal part of the limb bud and the bead was inserted into the limb mesoderm. We used heparin acrylic beads (Sigma, H5263) ranging between 100 and 200μm in diameter, soaked in BMP2(0.5μg/μl), BMP4(0.5μg/μl), Noggin (1μg/μl), FGF10(1μg/μl) or Gremlin (0.5 μg/μl) and ion exchange (AG1-X2, Bio-Rad) beads soaked in Wnt agonist (1μg/μl, Calbiochem), Wnt/β-catenin inhibitor (Cardamonin, 10mM) SU5402 (4 μg/μl, Calbiochem), retinoic acid (50μg/μl) or Citral (25% in DMSO, Fulka). As a control PBS-soaked beads were also implanted in all the experiments. The heparin acrylic beads were washed in PBS and incubated for 1 hour at room temperature in 2μl of the selected protein solution. For Wnt agonist, Wnt/β-catenin inhibitor and FGF inhibitor we used AG1-X2 beads and we followed a different protocol, preserving the molecules in the dark. For RA delivery, beads were added to a microtube with 50μl of the retinoid solution and were mixed for 30min. After that, the beads were spin down and the retinoic solution was substituted by 100μl of Dulbecco’s medium and mixed for 10min, twice. Finally the supernatant was removed and the beads were resuspended in 20μl of PBS.
After bead implantation, eggs were sealed with tape and re-incubated until embryos reached the desired range of development.
II.3.2 - In ovo electroporation
In ovo electroporation is a technique with a great potential that seems to be a very good
method to introduce exogenous DNA in eukaryotic cells by applying, in vivo, differential electric pulses in controlled conditions, using microelectrodes (Scaal et al., 2004). By applying these electric pulses, transient pores are formed allowing biological molecules injected in the
Materials and Methods
13 tissue to be transferred into cells, getting trapped there after the pores closure. (Rao et al., 2004; Sato et al., 2004; Scaal et al., 2004).
In this work, were electroporated embryos with 2 days of incubation (stage 14HH) which corresponds to the moment of development where the mesenchyme, that will form the hindlimb bud, become visible. Firstly, eggshell was swabbed with 70% ethanol and a small window was opened over the embryo. Then, the vitelline membrane above the limb field was carefully removed with fine tweezers and the right-dorsal surface of the lateral plate mesoderm was covered with the DNA, using a borosilicate glass capilar Kwik Fil™- WPI) coupled to a mouth pipette. Electrodes were placed over and under the prospective limb field in a parallel position to each other and to the neural tube with the negative electrode over the embryo and the positive one underneath (Figure 4). This way, DNA will be driven into the surface ectoderm of the prospective limb.
These electroporation experiments were performed with an Intracel TSS20 Ovodyne electroporator (Intracel LTD) using 3 pulses of 50 ms length (8 V each) with 60 ms interval. To avoid contamination, we added to each egg a drop of PBS/0.1% Pen/Strep and then they were sealed with tape and re-incubated. Finally, the incubated embryos were collected at different time points and fixed.
II.4 - Experimental construction of RNA interference (RNAi)
Introduction of double-stranded RNA (dsRNA) into a cell has proven to be a powerful tool for post-translational silencing of gene expression through a process known as RNA interference.
Materials and Methods
14 This is a highly conserved mechanism throughout taxonomical groups that was first observed in plants and fungi. In animals, RNA silencing was first reported in Caenorhabditis elegans and it is likely to also exist in humans (Elbashir et al., 2001; Tomari & Zamore, 2005). In addition to have an antiviral function, RNAi is also thought to be important to suppress the expression of potentially harmful segments of the genome, such as transposons, which could destabilize the genome (Duxbury & Whang, 2004; Tomari & Zamore, 2005).
After entering the cell, long dsRNAs are processed by RNAse III enzyme Dicer, which produces small interfering RNAs (siRNAs are 21-23 nucleotide dsRNA fragments with two nucleotide 3’ end overhangs). Then siRNAs are loaded into a ribonuclease known as RISC (RNA-induced silencing complex). It is the siRNAs that direct RISC to recognize mRNA containing a sequence homologous to the siRNA and cleaves the mRNA at the site located approximately in the middle of the homologous region.
There are a number of different strategies for inducing siRNA-mediated gene silencing. To perform RNAi experiments in chicken embryos, siRNAs were produced using pSUPER vector (OligoEngine, Inc.) that lead to loss-of-function phenotypes. This plasmid uses polymerase III H1-RNA promoter to express the forward sequence. Once this sequence is composed of small inverted repeats, separated by a spacer of three to nine nucleotides, short hairpin RNAs (shRNAs) will be formed, which are then processed by Dicer into siRNAs. After this, RISC will associate to those siRNAs. This way, RISC will cleave the target mRNA (the one that is complementary to the antisense strand of siRNAs), and once endogenous RNA can’t be totally transcribed, there is silencing of the target gene. Knockdown mediated by SUPER is maintained over long periods and its transcription products are not toxic to cells.
Materials and Methods
15
II.4.1 - RNAi cloning
Before transfection with pSuper plasmid we have to follow the following steps: select the target sequence and design primers, anneal these primers, phosphorylate oligos, dephosphorylate pSuper, ligate and transform E. coli.
Select the target sequence and design primers
Nowadays there are numerous online design tools that produce a list of suitable gene target sites. The designed target sequence represented in table 1, was obtained using the iRNAi program (http://www.mekentosj.com/irnai). This program supplies the target sequence based in the gene sequence that one want to silence, concerning some important considerations that we should have in mind: (1) it is usually recommended to chose a target site located at least 100-200 nucleotides from the AUG initiation codon, (2) 5’ and 3’ untranslated region (UTR) should ideally also be avoided, as associated regulatory protein could theoretically compromise RNAi; (3) 21-nucleotide siRNAs with 3’-d (TT) or (UU) overhangs are the most effective and most commonly used; (4) for optimal siRNA secondary structure, the GC ratio should ideally be between 45 and 55% and multiple identical nucleotides in series, particularly poly(C) and poly(G), should be avoided.
Table 1 -Primers for the synthesis of RNAi and its target sequence obtained for the clgr5
The design primers of 64 bp, are partially complementary between them and were purchased at MWG-Biotech AG (Ebersberg, Germany). Briefly, pSUPER vector was linearized with BglII and HindIII, forward and reverse strands of the oligos containing the siRNA-expressing sequence targeting lgr5 were annealed and cloned into the vector. The protocol followed for RNAi construct and its insertion in pSUPER vector is attached (Appendix III).
The plasmid was inserted by transformation into bacterial competent cells from Escherichia
coli strain DH5α (resistant to ampicillin; Invitrogen). The positive clones, transformants, should
show in an agarose gel a band of about 360 base pairs (bp) when the plasmid is double digested with EcoRI and HindIII, while clones without insert, unprocessed, should show only 300bp. FW 5’-GATCCCCCCCCGTGCACAATCTCCGCTTCAAGAGAGCGGAGATTGTCACGGGGTTTTTGGAAA-3’ RV 5’-AGCTTTTCCAAAAACCCCGTGCACAATCTCCGCTCTCTTGAAGCGGAGATTGTGCACGGGGGGG-3’ Target sequence AACCCCGTGCACAATCTCCGCTT GC content 56.5%
Materials and Methods
16
II.5 - Cloning of full length lgr5
II.5.1 - RNA extraction from tissuesThe RNA extraction of total tissue was made from whole chicken embryos, following the protocol of TRIZOL®Reagent – Invitrogen (see annex). TRIZOL®Reagent is a ready-to-use reagent that disrupts cells and dissolves cell components but maintain the integrity of RNA. After cellular lysis, addition of chloroform is followed by centrifugation, which separates the solution into an aqueous phase and an organic phase. RNA remains exclusively in the aqueous phase. After transferring the aqueous phase, the RNA is recovered by precipitation with isopropyl alcohol. In the end, the isolated RNA was precipitated and its concentration and purity was measured in a NanoDrop. All steps are performed under RNAse free conditions.
II.5.2 - 1st Strand DNA Synthesis
Preparation of cDNA was done by the 1st Strand Synthesis Kit for DNA RT-PCR (AMV-Virus Avian Myeloblastosis) according to the manufacturer's instructions (Fermentas). This kit allows reverse transcription of RNA into single stranded cDNA by the action of reverse transcriptase of AMV virus. The primers used for retrotranscription were random primers. See appendix III for detailed protocol.
II.5.3 -Amplification of the fragment to be cloned
The primers used for cloning the genes under study were ordered from the MWG
Oligo Synthesis Report (see table 2) and were used in a concentration of 10pmol/µL. The fragments were amplified by PCR (Polymerase Chain Reaction) from 100 ng of cDNA in PCR buffer (2mM dNTPs, 25mM MgCl2), and 0.5U of Taq DNA polymerase, in a final volume of 50µl. The mixture reaction was submitted to the following settings in a thermocycler:
PCR settings:
96 ˚C 4 min 96 ˚C 1 min
56 ˚C (annealing) 1.5 min 30 cycles 72 ˚C (extension) 2.5 min
72 ˚C 30 min 4 ˚C
These settings were adjusted for the cloning of larger products, where the elongation step must be increased 1 minute for every 1000 base pairs.
Materials and Methods
17 At the end, PCR product was analyzed in a 1% agarose gel and the correct band was extracted.
Table 2- Primers used to amplify the full length of clgr5 and its annealing temperatures
II.5.4 - Extraction and purification of a band in Agarose gel
For extraction and purification of PCR product from the agarose gel we used the QIAquick Gel
Extraction Kit (250) as instructed by manufacturer. This kit allows the extraction and
purification of DNA fragments from 70 bp to 10 kb.
The principle of this method is based in the fusion of agarose and selective adsorption of DNA molecules within particular size ranges by the silica membrane in the presence of high-salt buffer. Desorption of DNA was achieved with a solution with low concentration of salts like water. The effectiveness of this kit is up to 95%.
II.5.5 -Ligation
Purified PCR products, were ligated in the plasmid pGEM-Teasy (Promega), and the binding reaction was catalyzed by the enzyme T4 ligase. This vector, derived from the pGEM®-5Zf (+) (Promega), has at both 3' ends an additional Thymidine ideal for increasing the binding efficiency of PCR products to the plasmid because prevents vector recircularization and provides a terminal tail compatible with the originating by some thermostable polymerases. DNA of interest is inserted into the peptide coding for the enzyme β-galactosidase, a multiple cloning site (MCS) flanked by phage promoters (T7 and SP6), which can be used for in vitro production of RNA. In order to increase the efficiency of the ligation reaction, DNA of interest was used in a molar excess of 3 times over a cloning vector. The amount of DNA (ng of the insert) used for each ligation reaction can be calculated using the following formula:
The ligation reaction was left overnight at 4˚C. Animal model cDNA Primers pb TA Chicken lgr5 (full length) half part P1 Fw - 5’ ATGGCGACGTCCCGCGCTGACC 3’ 1472 56˚C … P2 Rv - 5’ TTGTAGTGGCTCTCGCAAGCTCC 3’ lgr5 (full lenght) 2nd half part P1 Fw - 5’ AAACTGGATGTGTCATCCAACC 3’ 1452 56˚C P2 Rv - 5’ CTAGCGACATGGAACAAATGC 3’
Materials and Methods
18
II.5.6 - Bacterial transformation
Transformation is the process by which the vector plasmid, including the cloned insert, is incorporated by competent bacteria in order to obtain a large number of recombinant clones transformed. For this, we added 5µl of ligation product to 50 µl of E. Coli DH5α competent cells, stored at -80°C and thawed slowly on ice. This mixture was placed on ice for 30 minutes and then for 30 seconds at 37°C in order to open transiently pores in the membrane allowing the entry of the bacterial plasmid. Finally, it was placed 2 minutes at 4°C to close the pores.In aseptic conditions were added 500 µl of liquid LB medium, and incubated for 1 hour in a dry bath at 37°C with agitation (300 rpm). Then, it was plated on solid LB medium with ampicillin and incubated overnight in Petri dishes at 37°C to allow the growth of colonies resistant to antibiotic selection. The plates were removed from the incubator at 37°C, and stored at 4°C.
II.5.7 - Transformants analysis
Isolated colonies were minced and inoculated into 2 ml of liquid LB medium supplemented with the appropriate antibiotic (ampicillin) in order to proceed with amplification of clones (about 16 hours at 37°C with shaking). Then, an extraction of plasmid DNA and subsequent restriction analysis were made to confirm if we had positive clones (with insert).
II.5.8 -Extraction of plasmidic DNA – Minipreps
For extraction and purification of plasmidic DNA we used a Wizard®Plus Minipreps DNA
Purification System Kit from PROMEGA (cat#A1460) following manufacturer's instructions.
These protocols are simple methods and high-income allowing rapid isolation of plasmidic DNA, based on lysis of bacterial cells that enables the release of the vector. At the end, plasmidic DNA was quantified by spectrophotometry.
II.5.9 - Digestion of the Plasmid
After plasmidic DNA extraction, we proceeded to the restriction analysis in order to verify which clones had the insertion of the DNA of interest by digestion with a restriction enzyme unique to the vector. For that, 1µg of DNA of each clone was digested, in a bath at 37°C for 3 to 4hours, with the appropriate enzyme, NotI in this case, and its buffer. The samples were then analyzed in agarose gel 0.8%, and the considered positive clones were sent for sequencing.
Materials and Methods
19
II.5.10 - Sequencing
For sequencing of positive clones, we added to the PCR tubes 2μL of BigDye® terminator sequencing buffer (5X), 2μL BigDye® terminator ready reaction mix, 400ng of template DNA and 3.2pmol of primers (T3, T7 or SP6) and water till a final volume of 10µl. Then, these mixtures were subjected to the following PCR thermocycler program:
96 ˚C 1 min 96 ˚C 10 sec 50 ˚C 5 sec 25 cycles 60 ˚C 4 min 4 ˚C 2 h
Then, we proceeded to DNA precipitation. For this, the content of each tube was transferred to a 1.5 ml centrifuge microtube and incubated at room temperature for 30 minutes, followed by centrifugation at 4°C and 14,000 rpm for 30 minutes. Removed the supernatant and recovered the sediment, the pellet was then washed with 250µl of 70% ethanol. The samples were mixed and centrifuged again at 14,000 rpm at 4˚C for 15 minutes.
Finally, the supernatant was removed and pellet dried. The samples were sent to the IGC sequencing service. The sequencing results were compared with sequences available in the database, prepared by alignment using the BLAST program (http://www.ncbi.nlm.nih.gov/BLAST).
II.5.11 - Amplification of the amount of DNA – Midipreps
In order to amplify the amount of DNA obtained previously we grew 100µl from one of the tubes with the best score of homology in 100 ml of LB medium. Then, midipreps protocol was performed, in accordance with the instructions of manufacturer, using a Wizard ® Plus DNA Midipreps Purification of PROMEGA System (A7640).This DNA purification system provide a simple and reliable method for rapid isolation of plasmid DNA, based on lysis of bacterial cells that enables the release of the vector.
II.6 - In situ hybridization
II.6.1 - Riboprobe preparation for whole-mount In situ hybridization
Chicken ESTs were available at the laboratory cloned into bluescript plasmid. RNA probes were synthesized from those plasmids using the restriction and RNA polymerase enzymes that are described in the table below (table 3). First, the plasmid vector containing the cDNA of the gene of interest was linearized and purified. Linearization was done in two different settings in order to generate templates for both antisense (positive probe) and sense (negative control
Materials and Methods
20 probe) probes. After digestion the DNA was purified by phenol:chloroform extraction, and precipitated with ethanol. After drying, the pellet was resuspended in milliQ water.
Table 3- Probes used: linearization enzyme and polymerase required for transcription of antisense Probe.
probe reference enzyme Pol. for AS
lgr4 ChEST534m17 NotI T3
lgr5 ChEST999g16 NotI T3
lgr6 ChEST244m20 NotI T3
lgr7 ChEST489a19 NotI T3
fgf8 ChEST320b9 NotI T3
en-1 ChEST92p12 NotI T3
lmx-1 ChEST100c17 NotI T3
II.6.2 - Plasmid Linearization
For this step, we digested plasmidic DNA with a unique restriction enzyme in order to create a linear fragment. To obtain the template cDNA for probe transcription, we digested 10μg of the plasmidic DNA containing the cDNA of interest using 1μL of the appropriate restriction enzyme, 2μL of the respective enzyme buffer (10x) and milliQ water for a final volume of 20 μL. The reaction occurred for about 4 hours at 37:C and then the completed digestion was confirmed by running 1μL of digestion reaction in a 0.8% agarose gel, observing a single band with the total plasmid size. After that, the linear plasmid was purified by phenol:chloroform extraction and precipitated with ethanol.
II.6.3 - Agarose gel electrophoresis
Agarose was dissolved in 1X TAE, usually at a final concentration of 0.8%. Then, in order to visualize DNA by UV light, RedSafe was added in the concentration of 1:4 from the stock solution. Loading dye was added to each sample (1x final concentration) as well as a DNA ladder used to estimate the size of the DNA fragments. By action of an electric field the molecules of nucleic acids, charged negatively, migrate into the agarose matrix of the negative to positive. In this work, an electric current of 80-100V was applied to the gel immersed in 1X TAE buffer.
II.6.4 - Phenol:Chloroform extraction and RNA/DNA precipitation with ethanol
When the digestion reaction was completed we purified DNA by phenol-chloroform extraction what allows us to remove proteins from the nucleic acid solution. For this, milliQ water was used to make a final volume of 100μL to reduce the loss of DNA during the process and an
Materials and Methods
21 equal volume of phenol-chloroform was added. The sample was mixed by strong vortexing and centrifuged for 5min at 14000 rpm. Following centrifugation, the mixture separates into two phases and we transferred the upper aqueous phase, containing the DNA, into a clean microtube and we precipitated it with ethanol. For the precipitation step, we added 0.1 volumes of 3M sodium acetate (pH5.2) in the case of DNA or the same amount of lithium chloride for RNA samples and 2.5 volumes of absolute ethanol for 30min at -80:C (or for over-night at -20:C). The precipitated RNA/DNA was recovered by centrifugation at 14000rpm for 30min at 4:C. Then supernatant was discarded and the pellet was washed with 5 volumes of 70% ethanol by centrifugation at 14000rpm for 15min at 4:C. Finally, the supernatant was discarded again and the RNA/DNA pellet was briefly dry. The precipitated RNA/DNA was resuspended in 20μL of milliQ water and stored at -20:C.
II.6.5 - In vitro DIG-labelled anti-sense RNA probe transcription
Riboprobes were synthesized by in vitro transcription with an adequate RNA polymerase and a mixture of dNTPs that contains DIG-labelled dUTPs. The synthesis of DIG-labelled anti-sense RNA probes was carried out by in vitro transcription at 37°C for 2h 30 min in a 20 μl reaction. The reaction contained 1X transcription buffer, 20U of RNase inhibitor, 1X DIG RNA labelling mix (Roche), 20 U of the appropriate RNA polymerase and 1 μg of the linearized template. After the generation of the riboprobe, 1 μl of the mixture was run on an agarose gel to estimate the amount of the probe. After this, RNA was precipitated with ethanol, resuspended in 20 μl of milliQ water and stored at - 20°C.
II.6.6 - Whole-mount In situ hybridization
The in situ hybridization represents a unique technique in which molecular biological and histochemical techniques are combined to study the precise cellular localization of gene expression by detection of specific messenger RNA sequences (mRNA) within intact cells or tissues. It consists in the use of a single-stranded gene-specific RNA probe (riboprobe) labelled with an epitope, in this case digoxigenin (DIG). The riboprobe will only bind to its complementary mRNA transcripts, allowing the detection at the target gene expression sites. This allows great specificity considering that RNA/RNA hybrids are thermodynamically very stable and that hybridization is performed at stringent conditions (higher temperature and increased content of formamide in the hybridization buffer). The DIG-labeled RNA was detected by an anti-DIG antibody that is coupled to alkaline phosphatase (AP) and hybridization was visualized by a permanent dye that precipitates using BM-purple (Jin, L. and Lloyd, R.).
In situ hybridization was performed as described in appendix III andin this approach it was used several proteinase K digestion times, according to each stage of development (see table 4).