Gonçalo Manuel Poças
Dissertation presented to obtain the Ph.D degree in Biochemistry
Instituto de Tecnologia Química e Biológica António Xavier | Universidade Nova de LisboaOeiras,
December, 2015
Gonçalo Manuel Poças
Dissertation presented to obtain the Ph.D degree in Biochemistry
Instituto de Tecnologia Química e Biológica António Xavier | Universidade Nova de LisboaOeiras, December, 2015
ii
Title: Functional and genetic analysis of alpha-synuclein and huntingtin in
Drosophila melanogaster
Dissertation presented to obtain the Ph.D. degree in Biochemistry
Author: Gonçalo Manuel Poças
Cell Signaling in Drosophila Laboratory
Instituto de Tecnologia Química e Biológica
Universidade Nova de Lisboa
Av. da República
Estação Agronómica Nacional
2780-157 Oeiras
Portugal
Front cover picture: image obtained by confocal microscopy of a Drosophila
brain expressing alpha-synuclein (green) in the dopaminergic neurons marked
by a specific antibody for Tyrosine Hydroxylase (blue).
Copyright © 2015 by Gonçalo Manuel Poças
I declare that the work presented in this thesis, except where otherwise stated,
is based on my own research and it was supervised by Doctor Pedro Manuel
Domingos. The work was mainly performed in Instituto de
Tecnologia Química e Biológica, Universidade Nova de Lisboa.
I am grateful for the financial support provided by Fundação para a Ciência e
Acknowledgements ... xi
Summary ... xiii
Sumário ... xv
Thesis publications ... xvii
List of acronyms ... xviii
Chapter I ... 1
General Introduction 1.1.Human neurodegenerative diseases ... 3
1.2.Parkinson’s disease ... 8
1.2.1. Epidemiology, symptoms and general molecular features ... 8
1.2.2. α-Synuclein: a major player in Parkinson’s disease ... 14
1.2.3. The vulnerability of dopaminergic neurons in Parkinson’s disease ... 17
1.3.Huntington’s disease ... 21
1.3.1. Epidemiology, symptoms and general molecular features ... 21
1.3.2. Huntingtin: the monogenic cause of Huntington’s disease ... 22
1.3.3. Medium spiny neurons vulnerability in Huntington’s disease ... 25
1.4.Modeling Parkinson’s and Huntington’s diseases in Drosophila ... 27
1.5.Main goals ... 35
1.6. References ... 36
Chapter II ... 49
The subcellular localization and axonal transport of α-synuclein in a Drosophila model for Parkinson’s disease 2.1. Abstract ... 51
2.2. Introduction ... 52
2.3. Results 2.3.1. The wild-type form of α-syn accumulates in the synaptic terminals of Drosophila photoreceptors ... 56
2.3.2. The A30P mutant version of α-syn is mislocalized in the Drosophila photoreceptors ... 57
2.3.3. No differences were detected in the aggregation state of the wild-type and the A30P mutant version of α-syn ... 59
2.3.4. Identification of α-syn WT and A30P protein interactors by co-immunoprecipitation and mass spectrometry analysis ... 62
viii
Chapter III ... 93
The role of huntingtin N-terminal phosphorylation on its aggregation and toxicity in a Drosophila model for Huntington’s disease 3.1. Abstract ... 95
3.2. Introduction ... 96
3.3. Results 3.3.1. Cdc25 inhibitors prevent mutant huntingtin aggregation and regulate its phosphorylation ... 100
3.3.2. The genetic knockdown of Cdc25 in Drosophila dopaminergic neurons reduced the aggregation of mutant Htt ... 103
3.3.3. The phosphorylation of NT17 residues modulates the oligomerization and aggregation of mutant Htt in human H4 glioma cells and in Drosophila dopaminergic neurons ... 105
3.3.4. The dephosphorylation of NT17 residues induces motor dysfunction and life span decrease in Drosophila ... 109
3.4. Discussion ... 112
3.5. Material and Methods... 114
3.6. References ... 119
Chapter IV ... 125
Crosstalk between Parkinson’s and Huntington’s diseases: α-synuclein modifies mutant huntingtin aggregation and neurotoxicity in Drosophila 4.1. Abstract ... 127
4.2. Introduction ... 128
4.3. Results 4.3.1. Co-expression of mutant Htt and α-syn alters Htt aggregation pattern .. 130
4.3.2. Htt103Q-mCherry and α-syn-EGFP co-localize and co-aggregate in dopaminergic neurons and in photoreceptor ... 132
4.3.3. Htt103Q-mCherry and α-syn-EGFP are physically interacting ... 135
4.3.4. Co-expression of Htt103Q-mCherry and α-syn-EGFP produces premature and severe degeneration in the photoreceptors ... 137
4.3.5. Co-expression of Htt103Q-mCherry and α-syn-EGFP in the nervous system causes severe motor dysfunction and a decrease in life span ... 139
4.4. Discussion ... 142
Chapter V ... 153
Testing the potential therapeutic effect of mannosylglycerate in Drosophila models for Parkinson’s and Huntington’s diseases 5.1. Abstract ... 155
5.2. Introduction ... 156
5.3. Results 5.3.1. Expression of MGSD in Drosophila ... 160
5.3.2. The co-expression of MGSD reduced the ER stress levels in photoreceptors expressing ninaEG69D ... 162
5.3.3. MG was not detected in cellular extracts from Drosophila tissues expressing MGSD ... 164
5.4. Discussion ... 168
5.5. Material and Methods... 170
5.6. References ... 173
Chapter VI ... 177
Final discussion 6.1. Final discussion ... 178
I would like to express my sincere gratitude to everyone who directly or
indirectly helped me to complete this thesis.
To my supervisor, Pedro Domingos, for allowing me to join his group
in 2008 when I was a master’s student, introducing me to the wonderful world
of Drosophila. Thank you for teaching me all the basics of Drosophila
genetics and crucial tricks and for giving me the freedom of exploring it on
my own during my PhD studies. Thank you for supporting me in the critical
phases of my PhD, both at the scientific and personal levels.
To all the present and former members of the group “Cell Signaling in
Drosophila” at ITQB. I would like to thank in particular to Fátima Cairrão, for
guiding me in some of the molecular biology techniques used in the beginning
of my PhD, to Vania Rasheva, for the support with Drosophila genetics, to
Catarina Gaspar, for all the nice scientific discussions, friendship and
companionship and to Yolanda Afonso, for helping me in the last 6 months in
some of my PhD projects.
To my collaborators Federico Herrera and Tiago Outeiro, who strongly
contributed to make the work in this thesis possible. I am also grateful to
Maria Luísa Vasconcelos for being part of my PhD thesis committee, together
with Tiago Outeiro, and for all the good scientific input and also for providing
me some fly stocks which were extremely useful during my PhD studies.
I also acknowledge the Fundação para Ciência e a Tecnologia (FCT) of
Ministry for Education and Science of Portugal for the indispensable financial
support under my PhD grant SFRH/BD/61477/2009. To Instituto de
Tecnologia Química e Biológica (ITQB) from Universidade Nova de Lisboa
for giving me the opportunity of performing my PhD studies in a very good
and stimulating environment, at the technical and intellectual levels. To
Instituto Gulbenkian de Ciência (IGC) for allowing me to use its facilities,
xii
and Margarida Saramago for the support and funny moments spent together.
To all the people from the former Tiago Outeiro’s group UNCM at IMM,
especially to Joana Branco-Santos for collaborating with me in some of my
PhD projects, to Patricia Guerreiro for helping me with the mass spectrometry
experiments and to Leonor Fleming for all her support, nice scientific
discussions and friendship.
To Diego Hartmann and Marija Petkovic for the friendship and
companionship during this long journey. Thank you for all the lunches we had
together, for all the funny moments, good conversations, advises and for
supporting me throughout all the frustrations and less positive periods but also
for sharing in the joy of the successes. To Luís Miguel Oliveira, who even
from the other side of the Atlantic Ocean, finds a way of encourage and
support me.
Finally, to all my dear friends and family for the love and support, with
especial words to my mother and my father, who always supported and
believed in me in all stages of my life, to my grandmother Deolinda, to my
grandfather António Melo and to my brother. To my dear grandparents Maria
do Carmo and Carlos Poças, who must be very proud, wherever they are. To
Jaime for all his support and words full of positive energy. To Filipe Martins
and Alpio James Aquilina for their great friendship and companionship. To
Rui for the extremely good moments during this last year, making everything
look a bit easier.
Muito obrigado a todos!
“Learn from yesterday, live for today, hope for tomorrow.
The important thing is not to stop questioning.”
In this work we established new transgenic Drosophila models for
Parkinson’s (PD) and Huntington’s disease (HD), two incurable devastating
human neurodegenerative diseases, which strongly compromise the patients’
motor abilities. Our Drosophila models are based on the transgenic
overexpression of fluorescent-tagged versions of two human neuronal proteins
extensively associated with these neuropathologies, but whose exact biological function is still unknown: alpha-synuclein (α-syn) for PD and huntingtin (Htt) for HD.
Here, we investigated whether the subcellular localization of α-syn and the defective axonal transport of this protein is relevant to the development of
PD. Using a Drosophila model for PD based on the overexpression of
EGFP-tagged versions of the wild-type and the familiar A30P mutant form of human α-syn, we observed a differential subcellular localization of the two versions of α-syn in the photoreceptors: while wild-type α-syn was enriched in pre-synaptic terminals, α-syn A30P was distributed throughout the cytoplasm of the photoreceptors, both in cell bodies and axons. We have identified, by
immunoprecipitation and mass spectrometry, the specific neuronal proteins interacting with wild-type and A30P α-syn, and by knocking-down the genes of these proteins we identified three candidates as neuronal modulators of α
-syn’s axonal transport and subcellular localization: Tomosyn, Spaghetti
Squash, and Synaptotagmin 4.
We also studied the role of N-terminal phosphorylation of mutant Htt in
HD. We used our Drosophila model for HD, based on the overexpression of a
mCherry-tagged version of the N-terminal truncated form of mutant human
Htt, encoded by the exon 1 of Htt gene (Httex1). We analyzed the relative
contribution of the phosphorylation state of each phosphorylatable residue in
xiv
neurotoxicity, depending on the biological context. Our findings suggest that
single phosphorylation events at NT17 could be a more effective therapeutic
approach against HD.
Taking advantage of our newly established Drosophila models for PD
and HD, we also investigated the possible crosstalk between these two neuropathologies, by studying the interaction of α-syn and Htt, at the genetic and functional levels. We showed that α-syn and mutant Htt co-aggregate in vivo when co-expressed in Drosophila and produce a synergistic
age-dependent increase in neurotoxicity associated to a decline in motor function and life span. Our results suggest that the co-existence of α-syn and Htt in the same neuronal cells worsens aggregation-related neuropathologies,
accelerating the disease progression.
Finally, we wanted to test the therapeutic properties of
mannosylglycerate (MG) in our Drosophila models for PD and HD. The
biosynthesis of MG can be catalyzed by MG synthase (MGSD). We
successfully generated transgenic lines expressing MGSD, but we could not
detect the biosynthesis and accumulation of MG in Drosophila.
We hope this work will contribute for a better understanding of the
molecular and cellular bases of PD and HD and that our new Drosophila
models for these pathologies may constitute one more useful platform
Neste trabalho estabelecemos novos modelos transgénicos de
Drosophila para as doenças de Parkinson (DP) e de Huntington (DH), duas
doenças humanas neurodegenerativas devastadoras e incuráveis que afectam
significativamente o controlo motor dos doentes. Os nossos modelos de
Drosophila baseiam-se na sobrexpressão de duas proteínas neuronais
humanas associadas a estas neuropatologias e para as quais a função biológica é desconhecida: alpha-synuclein (α-syn) para a DP e huntingtin (Htt) para a DH.
Um dos nossos objectivos consistia em investigar se a localização sub-celular da α-syn e o anormal transporte axonal desta proteína são relevantes no desenvolvimento da DP. Utilizando um modelo de Drosophila para a DP, baseado na sobrexpressão das formas “wild-type” e mutante A30P da α-syn fundidas com a “tag” fluorescente EGFP, observámos que o fenótipo relativo à localização sub-celular da α-syn é distinto para as duas formas da α-syn: a forma “wild-type” localiza-se predominantemente nos terminais
pré-sinápticos, enquanto que a mutante A30P distribui-se por todo o citoplasma
dos fotoreceptores, tanto nos corpos celulares como nos axónios. Através de
imunoprecipitação e espectrometria de massa, foi possível identificar as
proteínas que interagem especificamente com as versões “wild-type” e
mutante da α-syn e através do “knocking-down” dos genes que codificam para
estas proteínas conseguimos identificar três candidatos a moduladores da localização sub-celular e do transporte axonal da α-syn: Tomosyn, Spaghetti Squash, and Synaptotagmin 4.
Também estudámos o papel da fosforilação da porção N-terninal da Htt
mutante na DH. Para isso, utilizámos o nosso modelo de Drosophila para a
DH, baseado na sobrexpressão de uma versão truncada da porção N-terminal
da Htt mutante, codificada pelo exão 1 do gene da Htt (Httex1). Assim, foi
xvi
que eventos únicos de fosforilação no NT17 e fosfatases especificas modulam
efectivamente os níveis de agregação e neurotoxicidade da Htt mutante, de
uma forma que é dependente do contexto biológico. Assim, os nossos
resultados sugerem que eventos únicos de fosforilação no NT17 poderão
constituir uma estratégia terapêutica mais eficaz contra a DH.
Tirando partido dos novos modelos de Drosophila para as DP e DH
estabelecidos neste trabalho, o possível “crosstalk” entre estas duas doenças
foi também investigado, através do estudo da interacção genética e funcional
da α-syn e Htt. Desta forma, demonstrámos que a α-syn e a Htt mutante,
quando co-expressas em Drosophila, co-agregam “in vivo” e aumentam, de
uma forma sinergística e dependente da idade, a neurotoxicidade, disfunções
motoras e mortalidade. Assim, os nossos resultados sugerem que a co-existência da α-syn e Htt nas mesmas células neuronais poderá exacerbar as neuropatologias relacionadas com a agregação de proteínas, podendo acelerar
a progressão destas doenças.
Por último, também pretendiamos testar as propriedadades terapêuticas
do manosilglicerato (MG) nos nossos modelos de Drosophila para as DP e
DH. A biosíntese do MG pode ser catalizada pela MG sintetase (MGSD). Foi
possível produzir linhas transgénicas a expressar MGSD, mas não
conseguimos detectar a biosíntese e acumulação de MG em Drosophila.
Esperamos que este trabalho possa contribuir para um melhor
conhecimento das bases moleculares e celulares das DP e DH e que os nossos
novos modelos de Drosophila para estas patologias constituam mais uma
α-Synuclein modifies mutant Huntingtin aggregation and neurotoxicity in Drosophila. Human Molecular Genetics, 2015, 24(7):1898-907. DOI:
10.1093/hmg/ddu606
Manuscripts in preparation
Branco-Santos J*, Poças GM*, Herrera F, Domingos PM, Giorgini F, and
Outeiro TF N-terminal phosphorylation modulates mutant huntingtin
aggregation and neurotoxicity. * Equal contribution.
xviii
AD – Alzheimer’s disease
BiFC – Bimolecular Fluorescence Complementation
CAG – Cytosine-adenine-guanine triplet repeats
CNS – Central nervous system
Co-IP – Co-immunoprecipitation
CS – Compatible solutes
DA – Dopaminergic
DMSO – Dimethylsulfoxide
EIF4G1 – Eukaryotic translation initiation factor 4-gamma 1
GAL4/UAS – GAL4-dependent upstream activating sequence
GFP – Green fluorescent protein
GO – Gene ontology
GWAS – Genome Wide Association Studies
HD – Huntington’s disease
Htt – Huntingtin
Httex1 – Exon 1 of Htt gene
LDH – Lactate dehydrogenase
Lrrk2 – Leucine-rich repeat kinase 2
MG – Mannosylglycerate
MGSD – Mannosylglycerate synthase
MSN – Medium spiny neurons
PD – Parkinson’s disease
PolyQ – Polyglutamine
PRR – Proline-rich region
PTMs – Post-translational modifications
RP – Retinitis pigmentosa
S13 – Serine 13
S16 – Serine 16
SNCA – alpha-synuclein gene (non A4 component of amyloid precursor)
SNpc – Substantia nigra pars compacta
T3 – Threonine 3
TLC – Thin layer chromatography
UAS – Upstream activating sequence
VPS35 – Vacuolar protein sorting 35 homolog
VTA – Ventral tegmental area
1.1.Human neurodegenerative diseases
Human neurodegenerative diseases are a heterogeneous group of
devastating age-dependent disorders for which there is no cure or effective
symptomatic treatments.
These neuropathologies are characterized by the selective and
progressive loss of specific neuronal cells. The clinical manifestations of
each disease are determined by the region of the brain that degenerates
(Table 1.1). Although most neurodegenerative disorders display an array
of neural symptoms, they may be grouped and categorized in two major
groups: movement disorders and dementias. Movement disorders are
mainly characterized by the loss of motor control, in the form of tremor,
chorea, akinesia, bradykinesia or ataxia, for example. Parkinson’s disease
(PD) and Huntington’s disease (HD) belong to this category, although they
frequently show cognitive deficits and psychiatric problems concomitant
to motor deficits [1]. Dementias are mainly characterized by a severe loss
of cognitive function, as it occurs in Alzheimer’s disease (AD),
fronto-temporal dementia or dementia with Lewy bodies (LB). However,
dementias can also be accompanied by motor symptoms [2].
4
The genes responsible for many of the existing neurodegenerative
diseases have been identified, and they can be genetically inherited in a
recessive or dominant manner, depending on the disease. However, a high
percentage of cases are sporadic, being their causes and the associated
risks unclear. This is especially true for PD and AD, which are the two
most frequent neurodegenerative disorders [4]. Approximately 95% of PD
cases appear without a family history or a specific mutation [5].
The misfolding, aggregation and accumulation of neuronal proteins
into protein inclusions constitutes a common hallmark to most human
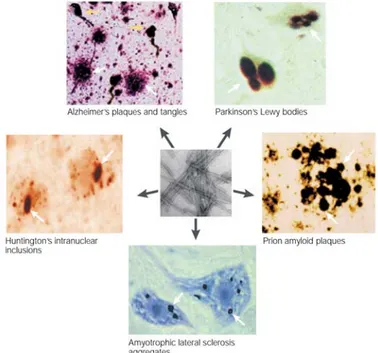
neurodegenerative diseases (Fig. 1.1).
Protein aggregates are visible in cells and tissues with basic
microscopic techniques, and still constitute the main post-mortem
diagnostic tool for neurodegenerative disorders, including PD [6, 7].
However, protein aggregation is a dynamic process that involves
seeding/nucleation mechanisms starting with the formation of small
soluble oligomeric species. This intermediate state of aggregation has a
high tendency to become stabilized by the formation of an oligomeric β-sheet structure, which by incorporation of additional monomers gives rise to protofibrils and finally to cross-β amyloid-like fibrils. Therefore protein oligomers constitute the nucleus material from which protein
aggregates start forming and growing, giving rise to the typical protein
amyloid-like inclusions observed in AD, PD and HD [8-10]. The generic term “amyloid” is commonly used to refer to the cross-β structure of the aggregates with binding affinities to Congo red and thioflavin S dies.
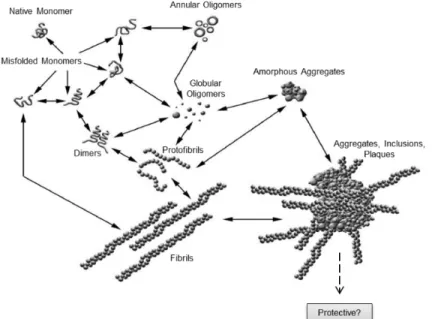
If large protein aggregates are the toxic entities responsible for
neurodegeneration or a protective response against other toxic species is
still widely debatable. There are several reports supporting a neurotoxic
role for fibrillary aggregates, in which large aggregates containing
misfolded proteins may interfere with the normal function of other
neuronal proteins, by sequestering them in these aggregates.[11-16].
However, the hypothesis that the large protein aggregates may
constitute a cellular defense mechanism against more toxic smaller
aggregates, or monomers, dimers, oligomers and protofibrils, has lately
6
Figure 1.2. Schematic representation of the process involved in the formation of amyloid inclusions in human neurodegenerative diseases. Several factors (such as, mutations, environmental stress and aging) may contribute to the misfolding and aggregation of native soluble monomer proteins. These aggregated monomers can adopt abnormal conformational structures, generating different intermediate species of aggregates (dimers, oligomers and protofibrils). A variety of complex pathways eventually give rise to large amyloid plaques, enriched in β-sheet fibrils, a histopathological hallmark of most human neurodegenerative diseases. These large aggregates were originally considered neurotoxic but accumulating evidences suggest they may have a cytoprotective role. Adapted from [17].
For example, in AD the cognitive impairment is not correlated with the density of Aβ plaques [18, 19], being the concentration of soluble Aβ much strongly correlated with the severity of the disease [20-22].
Equivalently, in PD LB could have a protective role, since the neurons
where these inclusions are mostly found are healthier than adjacent
neurons with no inclusions [23]. Soluble oligomeric species are often able
to cross membranes, move between cell compartments and outside the cell,
interacting with several macromolecules and consequently interfering with
The role of protein aggregates in neuropathologies remains poorly
understood and more studies are necessary to clarify if these aggregates
and the correspondent molecular mechanisms associated to protein
8
1.2. Parkinson’s disease
1.2.1.Epidemiology, symptoms and general molecular features
PD was firstly described in 1817 by James Parkinson as the
“shaking palsy” (Figure 1.3). The most prominent clinical symptoms are
related to motor functions: tremor, bradykinesia (slowness of movement),
rigidity and postural instability. In later stages, cognitive and behavioral
functions may also be affected. Post-mortem analyses of PD brains
revealed that PD is the second most frequent human neurodegenerative
disease after AD, affecting approximately 10 million patients worldwide.
Figure 1.3. First scientific description of Parkinson’s disease (PD) by James Parkinson in “An essay on the shanking palsy, 1817” and the illustration of this pathology by William Richard Gowers published in “A manual of diseases of the nervous system, 1886”.
PD is primarily an age-related disease and the number of affected
individuals is expected to increase significantly in the next decades as
PD, and neurodegenerative disorders in general, constitutes an
enormous socio-economic burden. For example, the current cost of the
treatments for patients with PD in United States is higher than 14 billion
dollars per year [26].
10
The presence of intraneuronal inclusions in surviving neurons,
called LB, constitutes one of the hallmarks of post-mortem brain analysis
of patients suffering from PD (Fig. 1.5). In PD the region of the brain most
affected by degeneration are the dopaminergic (DA) neurons from the
substantia nigra pars compacta (SNpc).
Figure 1.5. Lewy body (LB) and other cytoplasmic inclusions (CI) enriched in α -synuclein (α-syn) within the neurons of the dopaminergic neurons (DA) from substantia nigra pars compacta (SNpc) constitute an hallmark in Parkinson’s disease (PD). Extracted from [28].
PD is mostly sporadic, with familial forms constituting
approximately 10% of affected individuals. Eleven genes were associated
Table 1.2. Genetic loci associated to Mendelian Parkinson’s disease (PD). Extracted from [29].
Four of these loci are causative of autosomal dominant PD: SNCA,
encoding α-synuclein (α-syn); LRRK2, encoding leucine-rich repeat kinase
2; VPS35, encoding Vacuolar protein sorting 35 homolog and EIF4G1,
Eukaryotic translation initiation factor 4-gamma 1.
SNCA gene was the first to be identified as being associated to PD
and encodes α-syn, a neuronal protein of unknown function. Because α-syn is the principal component of the LB, it is considered a major player in this neuropathology. Several mutations have been mapped to SNCA
locus, including missense mutations and genomic duplications or
triplications, which account for the second most common cause of
dominant PD.
Mutations in LRRK2 are the most common genetic cause of
dominant PD, accounting for 10% of all familial forms. Lrrk2 is a
cytosolic protein with two predicted enzymatic domains, one with GTPase
and another with kinase activity. Although the exact biological function is
unknown, it has been associated with neurite formation and growth,
[30-12
34]. More recently, mutant versions of VPS35 and EIF4G1 were linked to
autosomal PD. VPS35 is one of the components of the tripartite complex
retromer, involved in the endosomal-lysosomal trafficking. The mutant
forms of VPS35 may affect the development of DA neurons through the
Wnt pathway [35, 36] andabnormal iron accumulation in the brain by the
DMT1 pathway [37, 38]. EIF4G1 is involved in mRNA translation [39].
Although the familial forms of PD constitute a minority, the
identification of PD loci has enabled the study and characterization of
some of the molecular mechanisms of idiopathic PD. For example, mutant
forms of Parkin, Pink 1 and F-box only protein 7 have been implicated in
the dysregulation of normal mitophagy [40-43], and mutations in the genes
encoding Lrrk2 and VPS35 may disrupt normal processes of protein
degradation by the autophagy/lysosomal pathway, ultimately leading to the
accumulation of cytotoxic misfolded proteins and cell death [44-47].
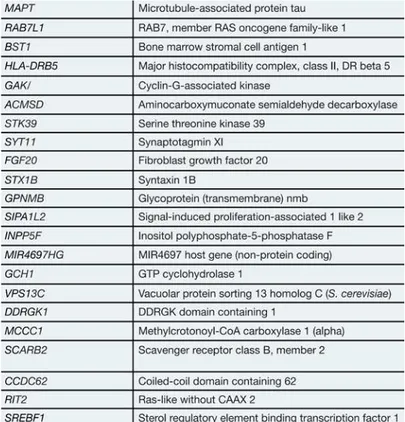
Besides the classical linkage analysis, genome-wide association
studies (GWAS) allowed to identify risk loci associated to sporadic forms
of PD (Table 1.3). However, the specific roles of these genes in the
molecular basis of PD remain unknown.
Table 1.3. Risk loci identified by several genome-wide association studies
(GWAS) as being associated with sporadic Parkinson’s disease (PD). Adapted
14
1.2.2 α-Synuclein: a major player in Parkinson’s disease
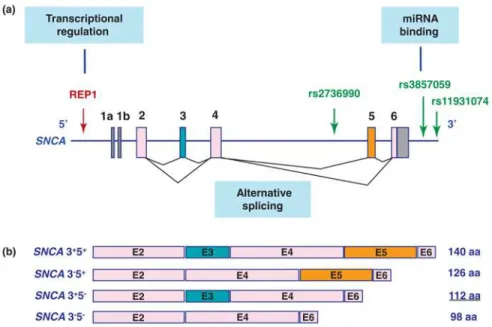
The synuclein family is constituted by α-, β- and γ-syn, but only α-syn has been found to be associated to human neurodegenerative diseases. α-Syn is a small neuronal protein composed by 140 amino acids, and encoded by the human SNCA gene in the chromosome 4 (Figure 1.6).
Figure 1.6. α-Synuclein (α-syn) is a major player in Parkinson’s disease pathogenesis. (A) α-syn is encoded by SNCA gene, located on chromosome 4. (B)
Several mutations segregated with PD have been already mapped to
the SNCA locus. The missense mutation A53T was mapped in 1997 [50].
Subsequently two additional missense mutations were mapped: A30P and
E46K [51, 52]. These missense mutations are located in the N-terminal
domain of α-syn, involved in the binding to membranes, and induce an
increase in the propensity of this protein to misfold and form fibrillar aggregates enriched in β-sheet structure [53-55]. More recently, the H50Q [56] and G51D [57] missense mutations were also identified. The H50Q
mutation stabilizes α-syn fibrils, significantly increasing the aggregation
rate and the ability of this protein to form amyloid inclusions, while the
G51D mutation slowdowns α-syn aggregation, but both mutations
increased the toxicity [58-61]. Genomic duplication or triplication of the SNCA locus, which increases α-syn expression, also causes a form of autosomal dominant PD [62-64]. The SNCA dosage is inversely correlated
with the age of onset and directly correlated with the severity of the
disease [65-67]. Post-mortem analysis of sporadic PD midbrain tissues revealed that the total mRNA levels for α-syn were significantly increased, when compared to control brains, further highlighting the relevance of the
levels of expression of this protein in PD [68].
GWAS indicated that variations at the SNCA locus are also strongly
correlated with the sporadic forms of PD, corroborating the importance of α-syn in the etiology of this neuropathology (Fig. 1.7). In fact, hundreds of single nucleotide polymorphisms (SNPs) within the SNCA locus have
been shown to increase susceptibility to PD (based on PDgene database – http://www.pdgene.org). Moreover, polymorphisms in the promoter region of SNCA have been also linked to PD [69]. The expansion of the
polymorphic dinucleotide repeat REP1 increases α-syn expression, thus
16
SNCA may also affect the stability and the alternative splicing of α-syn mRNA transcripts. Indeed, at least 6 different transcripts of this gene exist.
Besides the transcript encoding the full-length protein (140 aa), 5 truncated
forms have been described. One of these truncated forms (112 aa) has been
associated to LB formation and neurotoxicity [72].
Figure 1.7. Variations at the SNCA locus, identified by genome-wide association
studies (GWAS), associated to the sporadic forms of Parkinson’s disease (PD).
(A) The REP1 dinucleotide repeat, in the promoter region, regulates the levels of
SNCA expression; Three single nucleotide polymorphisms (SNP) that may alter
transcripts’ splicing and stability, and that are highly associated with PD, are also
indicated (in green). (B) Four isoforms of SNCA generated by alternative splicing
and implicated in the formation of Lewy bodies (LB) and neurotoxicity. Adapted
1.2.3 The vulnerability of dopaminergic neurons in Parkinson’s
disease
The specificity of the neuronal subpopulations that are mostly
affected and degenerate in each neurodegenerative disease raises a
fundamental question: are the insults specifically induced in those neurons
or are there specific cellular and molecular characteristics of the affected
neuronal subpopulations that render them more vulnerable to the insults?
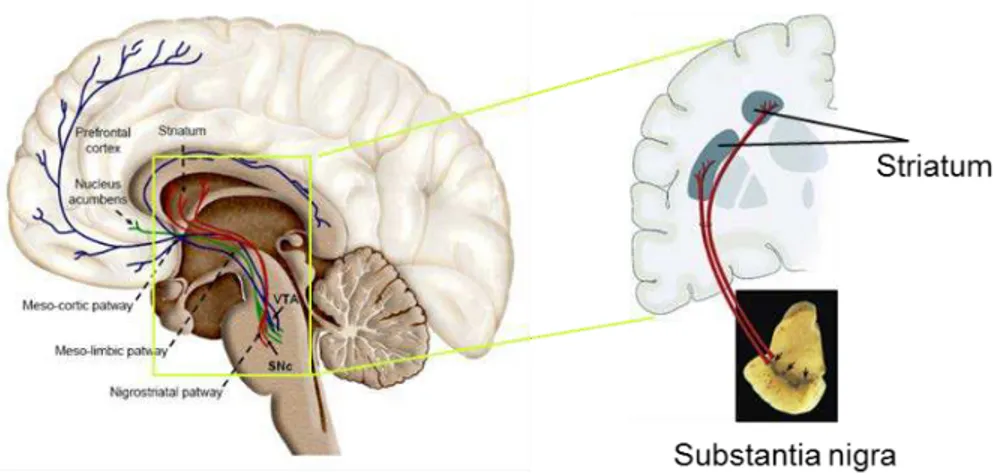
In the case of PD, the most affected neuronal population is the DA neurons
from SNpc that project their axons to the striatum (Figure 1.8).
Figure 1.8. The dopaminergic (DA) system and the nigrostriatal pathway. The DA neurons from substantia nigra pars compacta (SNpc), the most vulnerable neuronal population in Parkinson’s disease (PD), project their axons to the medium spiny neurons (MSN) from the striatum, constituting the nigrostriatal pathway, which plays a crucial role in the motor control.
The death of these neurons, and the consequent impairment of the
nigrostriatal DA pathway, is the cause of the motor symptoms observed in
PD. It is especially intriguing if we consider that from all the types of DA
18
neurons from ventral tegmental area (VTA), the DA neurons from SNpc
are the most vulnerable in PD.
Protein misfolding and aggregation, dysregulation of the ubiquitin
proteasome system, mitochondrial dysfunction and oxidative stress are not
exclusive of DA neurons, but are present in most areas of the PD brain
[73-75]. The difficulty in justifying the vulnerability of the DA neurons
from SNpc also persists when considering the familial forms of PD. The
expression of genes linked to this pathology is not specific and limited to
the DA neurons. For example, SNCA is ubiquitously expressed in the
central nervous system (CNS) at high levels, being also present in other
non-neuronal tissues [76].
The exposure to the neurotoxins 6-OHDA and MPTP specifically
induces neurodegeneration of the DA neurons from SNpc, and for this
reason they have been largely used as pharmacological models of PD. The
selectivity of these neurotoxins is explained by the fact that their
internalization into the cells is dependent on Sodium-dependent dopamine
transporter (DAT), which is only expressed in the DA neurons.
Importantly, DATis expressed at higher levels in SNpc than in the VTA
[77]. However, this explanation cannot be applied to other drugs that
induce PD-like pathology and symptoms, as is the case of the toxins
rotenone and paraquat, widely used as pesticide and herbicide,
respectively. Rotenone affects the mitochondrial function by inhibiting the
mitochondrial complex I, while paraquat induces oxidative stress, both
insults not being specifically targeted to the DA neurons from SNpc.
Bolan and Pissadaki [78] and by Brichta and Greengard [79] have
previously reviewed several particular determinants that put the SNpc DA
“on the edge” of the risk to degenerate in PD. Some of these
characteristics are related with the neuroanatomy of the nigrostriatal
are unmyelinated and extremely long, having a total cumulative length of
70 cm [80] and the estimated number of synapses established by these
axons is equally massive (200.000 – 400.000) [81, 82]. These unique
morphological features impose extreme bioenergetic demands, increasing
the metabolic needs of the SNpc DA neurons to maintain the membrane
potential, generate action potentials and enable synaptic transmission [83].
These exceptional energetic demands could make them especially
susceptible to insults of any sort, including environmental and genetic
factors [78].
Another important risk factor inherent to the DA neurons is the
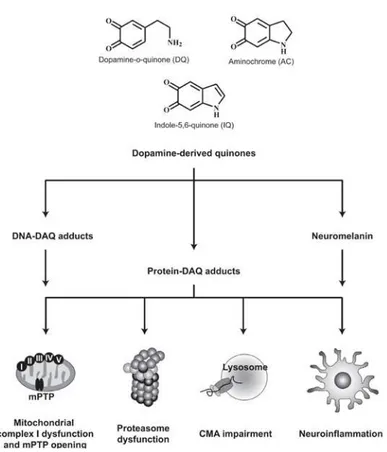
oxidation of dopamine and the consequent generation of reactive oxygen
species [84]. Dopamine is a potentially dangerous neurotransmitter within
the cells, as it easily undergoes oxidation generating dopamine-derived
quinones and other toxic molecules (Figure 1.9). Furthermore, cytoplasmic accumulations of misfolded α-syn may affect the secretory pathway by blocking the normal ER-Golgi traffic and thus exacerbating the deleterious
effects induced by toxic molecules, namely dopamine-derived products
20
Figure 1.9. Schematic representation of the molecular mechanisms associated to the toxic properties of the dopamine-derived quinones (DAQ). Extracted from [86].
The investigation of the specific cellular and molecular determinants
of the differential susceptibility of the DA neurons from SNpc in PD has
proven to be very challenging but holds great potential for the discovery of
1.3.Huntington’s disease
1.3.1. Epidemiology, symptoms and general molecular features
HD is the most common genetically inherited neurodegenerative
disease, belonging to the group of polyglutamine (polyQ) pathologies and
affecting approximately 5 to 10 per 100.000 individuals in the Caucasian
population [87-89]. It is caused by a mutation in the first exon of the
unconventionally large IT-15 gene (67 exons), which encodes a protein
called huntingtin (Htt).
Mutant Htt misfolds and accumulates into amyloid-like
proteinaceous aggregates in the medium spiny neurons (MSN) from the
striatum, being the most common post-mortem histopathological feature
(Fig. 1.10).
Figure 1.10. Post-mortem analysis of patients’ brains with Huntington’s disease (HD) show intranuclear inclusions (INI) and cytoplasmic inclusions (CI) containing mutant Huntingtin (Htt) accumulated. Extracted from [28].
Neurons from the thalamus, hippocampus and cortex also
degenerate, accounting for the typical symptoms observed in patients:
involuntary movements (chorea), dementia and psychiatric problems
(depression, anxiety, irritability, sexual dysfunction and suicidal
22
1.3.2. Huntingtin: the monogenic cause of Huntington’s disease
Since 1993, when the Htt gene was mapped to the short arm of
chromosome 4, intense research has been conducted aiming to identify the
biological function of Htt, as well as its role in the HD pathogenesis.
The disease-associated mutation of Htt occurs in the N-terminal
region of Htt and consists of an abnormally high number of
cytosine-adenine-guanine (CAG) triplet repeats. The expanded CAG repeats are
translated to an extended stretch of glutamines commonly called polyQ
tract [90, 91]. Htt is an unusually large protein, constituted by 3144 amino
acids (approximately 350 kDa). However, the N-terminal portion of
mutant Htt (exon 1) harboring the polyQ tract (Fig. 1.11) is sufficient to
produce HD phenotypes in vivo [92-97]. For this reason, most of the model
systems established to study HD in vitro and in vivo are based on the
expression of mutant versions of the exon 1 of Htt gene (Httex1).
The polyQ tract of wild-type Htt contains between 6 and 29
glutamines, while mutant alleles associated to HD contain over 36
glutamines. The longest polyQ tract ever detected in an HD patient was
constituted by 130 glutamines [98]. Individuals carrying intermediate
alleles containing a range of 29-35 glutamines are healthy and totally
asymptomatic. However, due to the meiotic instability of the CAG repeats,
these individuals have a very high probability to transmit a pathological
polyQ expansion over 36 glutamines to their offspring, especially from the
paternal side [99, 100].
The number of glutamines, which is responsible for approximately
70% of the variance, is inversely correlated with the age of onset of the
disease, with longer polyQ tracts inducing an earlier and more severe onset
of the pathology [101]. The mean age of HD onset is 38 years, although it
may vary between the ages of 25 and 70 years. PolyQ tracts containing
over 55 glutamines (5% of all cases) produce Juvenile HD, where the onset
can start before the age of 20 years [102]. The mean duration of the disease
is 17-20 years and patients usually die from pneumonia or suicide [103].
Despite the efforts made to date, there is still no cure for HD and the
drugs available can only treat the symptoms. For the development of
effective treatments for HD, it is essential to understand the cellular and
molecular mechanisms underlying this pathology.
The exact biological function of Htt is unknown, but it was shown to
possess an indispensable anti-apoptotic role [104, 105]. There is also
evidence that Htt is essential for normal development, as Htt-null mutant
mice die as embryos at day 7.5 [104, 106, 107]. Surprisingly, Htt is
dispensable for Drosophila development, but it is crucial for normal
long-term mobility and survival in adult flies [108]. Htt interacts with a high
24
functions, such as endocytosis, neurotransmission, transcriptional
regulation, axonal transport and apoptosis [109-111]. Htt could act as
scaffold, responsible for bringing together its protein partners and for
coordinating the transfer of information among different subcellular
compartments, namely between the nucleus and cytoplasm [111]. Htt
could also act as a scaffold protein in selective autophagy by promoting
cargo recognition and autophagy initiation [112].
The expansion of the polyQ tract promotes conformational changes
in Htt that dramatically increase its propensity to misfold and aggregate.
Htt aggregation could affect the interaction with its neuronal partners and
disturb the normal function of Htt and Htt-interacting proteins. This
suggests the involvement of both toxic gain-of-function and
loss-of-function mechanisms in the development of HD, by disrupting these
1.3.3. Medium spiny neurons vulnerability in Huntington’s disease
Although Htt is ubiquitously expressed throughout nervous system
and other peripheral tissues (namely, testes, liver, heart and lungs) the
pathological effects are predominantly induced in specific neuronal
populations of the brain, especially the MSN neurons from the striatum
[113-118]. The fact that mutant Htt affects this particular neuronal
population points to the possible relevance of cell- or tissue-specific
factors in HD pathogenesis, beyond the expanded polyQ tract.
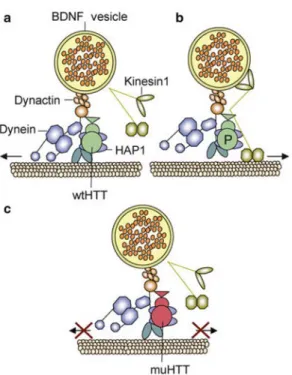
Brain-derived neurotrophic factor (BDNF) is a possible determinant
of the increased susceptibility of MSN neurons to neurodegeneration in
HD. BDNF is a neurotrophin necessary for the development, survival and
proper function of the striatal neurons. Deficient BDNF signaling in the
striatum, presumably caused by mutant Htt, has been pointed as strong
candidate to be a molecular determinant and modulator in HD
26
1.4. Modeling Parkinson’s and Huntington’s diseases in Drosophila
The establishment of animal models is one of the most useful
approaches to study the pathogenic mechanisms of human diseases at the
molecular, cellular, organic and functional level. Drosophila
melanogaster, commonly known as fruit fly, is a powerful genetic model
organism that has been used to study complex biological phenomena for
more than a century, including human neurodegenerative diseases [121].
The identification of genes associated to the familial forms of PD
and HD in the last decade allowed the establishment of animal models for
these pathologies, which are essential to investigate the early
pre-symptomatic stages of pathogenesis and to test new drug candidates.
One of the key factors that contributed to the great success of
Drosophila as a model organism to study human pathologies is the very
high degree of conservation with mammals, since approximately 75% of
disease-related loci in humans have at least one Drosophila homologue
[122]. Drosophila also offers a great number of potent genetic tools, it has
a very well-known anatomy and has short life cycle and life-span. Finally,
the central nervous system of invertebrates and vertebrates shares a
common evolutionary origin. Flies have a complex nervous system
capable of learning and coordination of intricate behaviors, and there is a
significant degree of genetic and functional conservation between the fly
central complex and the human basal ganglia, which is the region
primarily affected in PD and HD [123]. Such homology constitutes an
undeniable advantage to model and study these and other human
neurodegenerative diseases.
In order to model a human neurodegenerative disease in flies, it is
necessary to express in this organism the human genes associated with this
28
Drosophila is by using the binary GAL4-dependent upstream activating
sequence (GAL4/UAS) system [124] (Fig. 1.13).
Figure 1.13. The GAL4/UAS system, used for overexpression of proteins in
Drosophila, is a binary system which allows for the ectopic expression of genes of
interest in a specific tissue or cell type. Two transgenic fly lines are created: the UAS line, in which the gene of interest is placed downstream of a UAS (Upstream Activating Sequence) domain, where the yeast transcriptional activator GAL4 binds; and the GAL4 line (driver), which expresses Gal4 under the control of a tissue specific promoter. The gene encoded in the UAS line is only activated when this line is crossed with the GAL4 line. In our study we generated UAS lines
encoding for α-synuclein (α-syn) and huntingtin (Htt) and we used pan-neuronal, dopaminergic and photoreceptor drivers.
The gene of interest is subcloned into an UAS expression construct,
which is microinjected into fly embryos to establish transgenic lines. The
protein of interest is expressed in a targeted way by performing the genetic
cross of the UAS line with a Gal4 line that expresses the yeast
transcriptional co-activator GAL4 in a specific tissue or cell type.
the presence of GAL4 in cells and tissues. Many cell-type and
developmentally regulated GAL4 lines, commonly called “drivers”, are
readily available from Drosophila public stock centers (e.g. Bloomington
Drosophila Stock Center at Indiana University). So, the effect of
expressing a human transgene in many different tissues and at various
developmental stages can be assayed without creating many independent
transgenic lines. The GAL4/UAS system is especially useful when one
aims to study the mechanisms of toxicity of human genes linked to the disease which are absent in the Drosophila genome, as is the case of α-syn.
In the first Drosophila model of PD, transgenic flies expressing wild-type or two familial mutant forms (A30P and A53T) of human α-syn in the brain reproduced key features of PD, including LB-like inclusions
(Fig. 1.14), selective degeneration of DA neurons, and abnormalities in the
locomotor behavior [125]. Actually, Drosophila reproduced PD
phenotypes better than mice models of the disease.
30
However, in another report, missexpression of α-syn in the Drosophila CNS did not cause death of the DA neurons [127]. The
inconsistency of the results from these independent studies may result
from the use of different technical approaches. Therefore, there is the need
for more independent studies in order to clarify some of these
inconsistencies and to generate Drosophila models that consistently
reproduce the key phenotypes resembling PD conditions.
Concerning HD, Drosophila models show the essential features
associated to the pathology, such as progressive neurodegeneration [128],
motor deficits, protein inclusions in cytoplasm and nucleus (Figure 1.15)
and a correlation between polyglutamine repeat length, age of onset and
severity of the phenotypes [129].
In this work we have established new transgenic Drosophila models
for PD and HD, based on the overexpression of wild-type and mutant versions of α-syn and Htt (Fig. 1.16).
Figure 1.16. We generated UAS transgenic lines encoding fluorescent-tagged versions of two human proteins associated with Parkinson’s (PD) and
Huntington’s diseases (HD). For PD we generated constructs of human α
-synuclein (α-syn) fused to EGFP and we used a wild-type (WT) and a familiar mutant form (the missense mutation A30P) of this protein. For HD we generated mCherry-tagged versions of a wild-type form with 19 glutamines (19Q) and a mutant form containing 97 glutamines (97Q) of human huntingtin (Htt).
For the PD model, we used the missense mutation A30P, associated
to familial cases of the disease; and for the HD model, we used wild-type
and mutant forms of Htt, with 19Q and 97Q in the polyQ tract, respectively. These proteins were fused to fluorescent tags (EGFP for α-syn and mCherry for Htt), allowing microscopy and co-localization
studies in either “live” or fixed biological materials.
Taking advantage of the Gal4/UAS system, we were able to induce
targeted expression of these proteins in different neuronal tissues of
interest, such as in the whole nervous system, using the pan-neuronal
driver Elav-Gal4, in the eye retina, using the sGMR-Gal4 driver and in the
32
Figure 1.17. Targeted expression of EGFP-tagged α-synuclein using the Gal4/UAS system. Confocal microscopy images of adult flies expressing α -synuclein-EGFP in different tissues: (A) in the whole-brain, using the pan-neuronal driver Elav-Gal4; (B) in the photoreceptors, using the GMR-Gal4 driver; (C) in the dopaminergic (DA) neurons, using the TH-Gal4 driver.
Throughout our study, depending on the specific questions and on
the particular experiments to be done, we induced the targeted expression
of the proteins of interest in different tissues, such as the whole brain, the
eye photoreceptors or the DA neurons. The DA neurons constituted one of
our favorite neuronal populations to study PD and HD in our Drosophila
models, being the TH-Gal4 driver extensively used in our study. This
driver expresses Gal4 under the control of the promotor region of tyrosine
hydroxylase (TH) gene. TH is the enzyme that catalyzes the rate-limiting
step of dopamine biosynthesis and is specifically expressed in all DA cells.
The DA neurons, besides being the neuronal cell type directly
affected in PD, the axons of the DA neurons constitute the main input to
the MSN from the striatum, affected in HD. The DA system in mammals is
involved in the control of locomotor behavior, motivational states and
cognitive function, all of them impaired to different degrees in PD and
HD. Furthermore, the impairment of particular behaviors, such as the
initiation of voluntary actions in mammals has been correlated with the
malfunction of specific subpopulations of DA neurons, namely the ones
Drosophila DA system is well characterized by means of dopamine
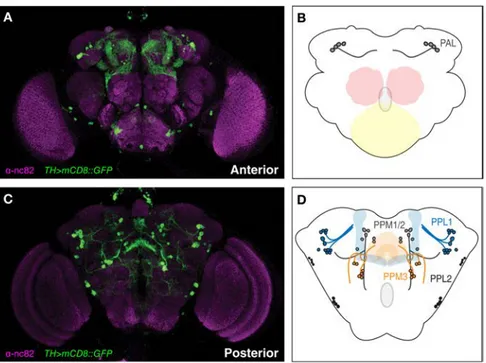
and anti-Tyrosine Hydroxylase (TH) immunoreactivity, enabling the
characterization of several DA clusters which were named according to
their anatomical localization in the brain: paired posterior lateral 1 and 2
(PPL1 and PPL2); paired posterior medial 1 and 2 (PPM1/2); paired
posterior medial 3 (PPM3); paired anterior lateral (PAL), and paired
anterior medial (PAM). (Fig. 1.18) [132-134]. Similarly to mammals,
Drosophila DA system is also involved in the modulation and control of
locomotor behavior. Furthermore, specific subset of DA neurons are
especially vulnerable to neurodegeneration and to induce motor deficits in
the context of PD, which is equivalent to the situation observed in humans
[135, 136].
For these reasons, we consider Drosophila DA neurons a very
useful and adequate system to model and study human neurodegenerative
34
1.5. Main goals
Using our newly established Drosophila models, we were interested
in the study of three different particular aspects that we believe to be
relevant to the molecular pathogenesis of PD and HD:
the molecular determinants of the subcellular localization of α-syn
in PD (Chapter II)
the effect of N-terminal phosphorylation in the aggregation and
toxicity of Htt in vitro and in vivo (Chapter III)
the possible crosstalk between PD and HD molecular mechanisms,
by studying the genetic and functional interaction between α-syn
and Htt (Chapter IV)
Additionally, we tried to identify new potential therapeutic
compounds for PD and HD, being particularly interested in the putative
therapeutic effect of mannosylglycerate (MG) in our Drosophila models of
neurodegeneration (Chapter V). MG is a compatible solute, i.e. a substance
compatible with the cellular metabolism that accumulates in the cytoplasm
to balance external osmotic pressure. These properties confer potential
neuroprotective effects to this compound in the context human
neurodegenerative diseases, as previously shown by our collaborators
using a yeast model for PD [137].
We believe this work will contribute to a better understanding of the
molecular and cellular pathophysiology mechanisms underlying PD and
36
1.6. References
1. Perry, R.J. and J.R. Hodges, Spectrum of memory dysfunction in degenerative disease. Curr Opin Neurol, 1996. 9(4): p. 281-5. 2. Tsolaki, M., et al., Extrapyramidal symptoms and signs in
Alzheimer's disease: prevalence and correlation with the first symptom. Am J Alzheimers Dis Other Demen, 2001. 16(5): p. 268-78.
3. Soto, C., Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci, 2003. 4(1): p. 49-60.
4. Nussbaum, R.L. and C.E. Ellis, Alzheimer's disease and Parkinson's disease. N Engl J Med, 2003. 348(14): p. 1356-64.
5. van der Vegt, J.P., et al., Imaging the impact of genes on Parkinson's disease. Neuroscience, 2009. 164(1): p. 191-204. 6. Hughes, A.J., et al., The accuracy of diagnosis of parkinsonian
syndromes in a specialist movement disorder service. Brain, 2002. 125(Pt 4): p. 861-70.
7. Goedert, M., et al., 100 years of Lewy pathology. Nat Rev Neurol, 2013. 9(1): p. 13-24.
8. Jarrett, J.T., E.P. Berger, and P.T. Lansbury, Jr., The C-terminus of the beta protein is critical in amyloidogenesis. Ann N Y Acad Sci, 1993. 695: p. 144-8.
9. Scherzinger, E., et al., Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington's disease pathology. Proc Natl Acad Sci U S A, 1999. 96(8): p. 4604-9.
10. Wood, S.J., et al., alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson's disease. J Biol Chem, 1999. 274(28): p. 19509-12.
11. Lorenzo, A. and B.A. Yankner, Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci U S A, 1994. 91(25): p. 12243-7.
12. Pike, C.J., et al., Neurodegeneration induced by beta-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci, 1993. 13(4): p. 1676-87.
14. Kayed, R., et al., Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Abeta oligomers. Mol Neurodegener, 2010. 5: p. 57. 15. Okada, T., et al., Formation of toxic fibrils of Alzheimer's amyloid
beta-protein-(1-40) by monosialoganglioside GM1, a neuronal membrane component. J Mol Biol, 2007. 371(2): p. 481-9.
16. Novitskaya, V., et al., Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons. J Biol Chem, 2006. 281(19): p. 13828-36.
17. Lotz, G.P. and J. Legleiter, The role of amyloidogenic protein oligomerization in neurodegenerative disease. J Mol Med (Berl), 2013. 91(6): p. 653-64.
18. Terry, R.D., et al., Some morphometric aspects of the brain in senile dementia of the Alzheimer type. Ann Neurol, 1981. 10(2): p. 184-92.
19. Hashimura, T., T. Kimura, and T. Miyakawa, Morphological changes of blood vessels in the brain with Alzheimer's disease. Jpn J Psychiatry Neurol, 1991. 45(3): p. 661-5.
20. Lue, L.F., et al., Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol, 1999. 155(3): p. 853-62.
21. McLean, C.A., et al., Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol, 1999. 46(6): p. 860-6.
22. Naslund, J., et al., Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA, 2000. 283(12): p. 1571-7.
23. Tompkins, M.M. and W.D. Hill, Contribution of somal Lewy bodies to neuronal death. Brain Res, 1997. 775(1-2): p. 24-9.
24. Agorogiannis, E.I., et al., Protein misfolding in neurodegenerative diseases. Neuropathol Appl Neurobiol, 2004. 30(3): p. 215-24. 25. Taylor, J.P., J. Hardy, and K.H. Fischbeck, Toxic proteins in
neurodegenerative disease. Science, 2002. 296(5575): p. 1991-5. 26. Kowal, S.L., et al., The current and projected economic burden of
Parkinson's disease in the United States. Mov Disord, 2013. 28(3): p. 311-8.
27. Dorsey, E.R., et al., Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030.
38
28. Ross, C.A. and M.A. Poirier, Opinion: What is the role of protein aggregation in neurodegeneration? Nat Rev Mol Cell Biol, 2005. 6(11): p. 891-8.
29. Spatola, M. and C. Wider, Genetics of Parkinson's disease: the yield. Parkinsonism Relat Disord, 2014. 20 Suppl 1: p. S35-8. 30. Winner, B., et al., Adult neurogenesis and neurite outgrowth are
impaired in LRRK2 G2019S mice. Neurobiol Dis, 2011. 41(3): p. 706-16.
31. Dachsel, J.C., et al., A comparative study of Lrrk2 function in primary neuronal cultures. Parkinsonism Relat Disord, 2010. 16(10): p. 650-5.
32. Tong, Y., et al., Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol Neurodegener, 2012. 7: p. 2.
33. Ren, Y., et al., Selective vulnerability of dopaminergic neurons to microtubule depolymerization. J Biol Chem, 2005. 280(40): p. 34105-12.
34. Piccoli, G., et al., LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J Neurosci, 2011. 31(6): p. 2225-37.
35. Castelo-Branco, G., et al., Differential regulation of midbrain dopaminergic neuron development by 1, 3a, and Wnt-5a. Proc Natl Acad Sci U S A, 2003. 100(22): p. 12747-52.
36. Port, F., et al., Wingless secretion promotes and requires retromer-dependent cycling of Wntless. Nat Cell Biol, 2008. 10(2): p. 178-85.
37. Deng, H., K. Gao, and J. Jankovic, The VPS35 gene and Parkinson's disease. Mov Disord, 2013. 28(5): p. 569-75.
38. Tabuchi, M., et al., Retromer-mediated direct sorting is required for proper endosomal recycling of the mammalian iron transporter DMT1. J Cell Sci, 2010. 123(Pt 5): p. 756-66.
39. Schulte, E.C., et al., Variants in eukaryotic translation initiation factor 4G1 in sporadic Parkinson's disease. Neurogenetics, 2012. 13(3): p. 281-5.
40. Kane, L.A., et al., PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol, 2014. 205(2): p. 143-53.
42. Koyano, F., et al., Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature, 2014. 510(7503): p. 162-6.
43. Lazarou, M., et al., The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature, 2015. 524(7565): p. 309-14.
44. Orenstein, S.J., et al., Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci, 2013. 16(4): p. 394-406.
45. Manzoni, C., et al., Inhibition of LRRK2 kinase activity stimulates macroautophagy. Biochim Biophys Acta, 2013. 1833(12): p. 2900-10.
46. Alegre-Abarrategui, J., et al., LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum Mol Genet, 2009. 18(21): p. 4022-34.
47. Beilina, A., et al., Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci U S A, 2014. 111(7): p. 2626-31.
48. Bras, J., R. Guerreiro, and J. Hardy, SnapShot: Genetics of Parkinson's disease. Cell, 2015. 160(3): p. 570-570 e1.
49. Venda, L.L., et al., alpha-Synuclein and dopamine at the crossroads of Parkinson's disease. Trends Neurosci, 2010. 33(12): p. 559-68.
50. Polymeropoulos, M.H., et al., Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science, 1997. 276(5321): p. 2045-7.
51. Kruger, R., et al., Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet, 1998. 18(2): p. 106-8. 52. Zarranz, J.J., et al., The new mutation, E46K, of alpha-synuclein
causes Parkinson and Lewy body dementia. Ann Neurol, 2004. 55(2): p. 164-73.
53. Coskuner, O. and O. Wise-Scira, Structures and free energy landscapes of the A53T mutant-type alpha-synuclein protein and impact of A53T mutation on the structures of the wild-type alpha-synuclein protein with dynamics. ACS Chem Neurosci, 2013. 4(7): p. 1101-13.
40
55. Lemkau, L.R., et al., Mutant protein A30P alpha-synuclein adopts wild-type fibril structure, despite slower fibrillation kinetics. J Biol Chem, 2012. 287(14): p. 11526-32.
56. Appel-Cresswell, S., et al., Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov Disord, 2013. 28(6): p. 811-3.
57. Kiely, A.P., et al., alpha-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson's disease and multiple system atrophy? Acta Neuropathol, 2013. 125(5): p. 753-69. 58. Rutherford, N.J., et al., Divergent effects of the H50Q and G51D
SNCA mutations on the aggregation of alpha-synuclein. J Neurochem, 2014. 131(6): p. 859-67.
59. Porcari, R., et al., The H50Q mutation induces a 10-fold decrease in the solubility of alpha-synuclein. J Biol Chem, 2015. 290(4): p. 2395-404.
60. Lesage, S., et al., G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol, 2013. 73(4): p. 459-71.
61. Fares, M.B., et al., The novel Parkinson's disease linked mutation G51D attenuates in vitro aggregation and membrane binding of alpha-synuclein, and enhances its secretion and nuclear localization in cells. Hum Mol Genet, 2014. 23(17): p. 4491-509. 62. Singleton, A.B., et al., alpha-Synuclein locus triplication causes
Parkinson's disease. Science, 2003. 302(5646): p. 841.
63. Muenter, M.D., et al., Hereditary form of parkinsonism--dementia.
Ann Neurol, 1998. 43(6): p. 768-81.
64. Konno, T., et al., Autosomal dominant Parkinson's disease caused by SNCA duplications. Parkinsonism Relat Disord, 2015.
65. Miller, D.W., et al., Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology, 2004. 62(10): p. 1835-8.
66. Ibanez, P., et al., Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet, 2004. 364(9440): p. 1169-71.
67. Chartier-Harlin, M.C., et al., Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet, 2004. 364(9440): p. 1167-9.
69. Pihlstrom, L. and M. Toft, Genetic variability in SNCA and Parkinson's disease. Neurogenetics, 2011. 12(4): p. 283-93.
70. Goldman, S.M., et al., Head injury, alpha-synuclein Rep1 and Parkinson's disease: a meta-analytic view of gene-environment interaction. Eur J Neurol, 2015. 22(7): p. e75.
71. Janeczek, P., et al., Reduced expression of alpha-synuclein in alcoholic brain: influence of SNCA-Rep1 genotype. Addict Biol, 2014. 19(3): p. 509-15.
72. Kalivendi, S.V., et al., Oxidants induce alternative splicing of alpha-synuclein: Implications for Parkinson's disease. Free Radic Biol Med, 2010. 48(3): p. 377-83.
73. Cookson, M.R., Parkinsonism due to mutations in PINK1, parkin, and DJ-1 and oxidative stress and mitochondrial pathways. Cold Spring Harb Perspect Med, 2012. 2(9): p. a009415.
74. Lee, F.J. and F. Liu, Genetic factors involved in the pathogenesis of Parkinson's disease. Brain Res Rev, 2008. 58(2): p. 354-64.
75. Shen, J. and M.R. Cookson, Mitochondria and dopamine: new insights into recessive parkinsonism. Neuron, 2004. 43(3): p. 301-4.
76. Ltic, S., et al., Alpha-synuclein is expressed in different tissues during human fetal development. J Mol Neurosci, 2004. 22(3): p. 199-204.
77. Lammel, S., et al., Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron, 2008. 57(5): p. 760-73.
78. Bolam, J.P. and E.K. Pissadaki, Living on the edge with too many mouths to feed: why dopamine neurons die. Mov Disord, 2012. 27(12): p. 1478-83.
79. Brichta, L. and P. Greengard, Molecular determinants of selective dopaminergic vulnerability in Parkinson's disease: an update.
Front Neuroanat, 2014. 8: p. 152.
80. Matsuda, W., et al., Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci, 2009. 29(2): p. 444-53.
81. Oorschot, D.E., Total number of neurons in the neostriatal, pallidal, subthalamic, and substantia nigral nuclei of the rat basal ganglia: a stereological study using the cavalieri and optical disector methods. J Comp Neurol, 1996. 366(4): p. 580-99.
82. Arbuthnott, G.W. and J. Wickens, Space, time and dopamine.