J. Braz. Chem. Soc. vol.28 número11
Texto
Imagem
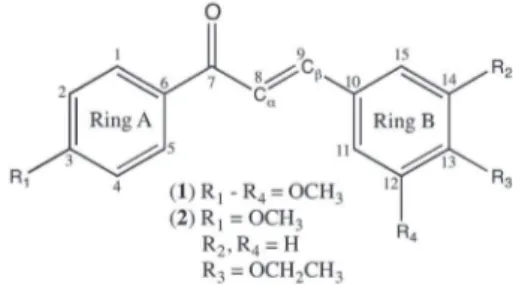
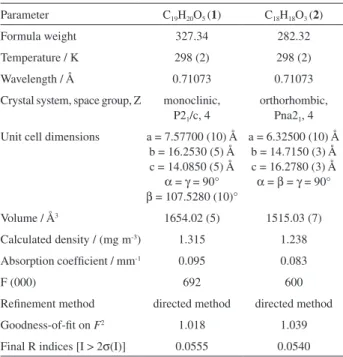
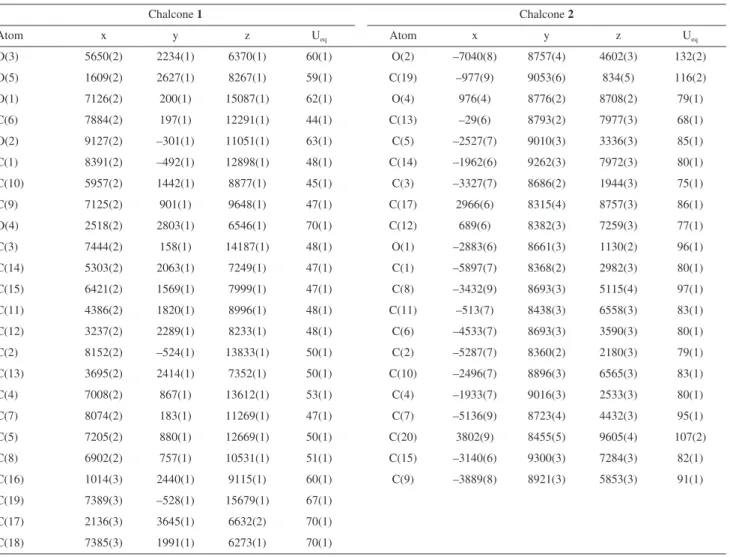
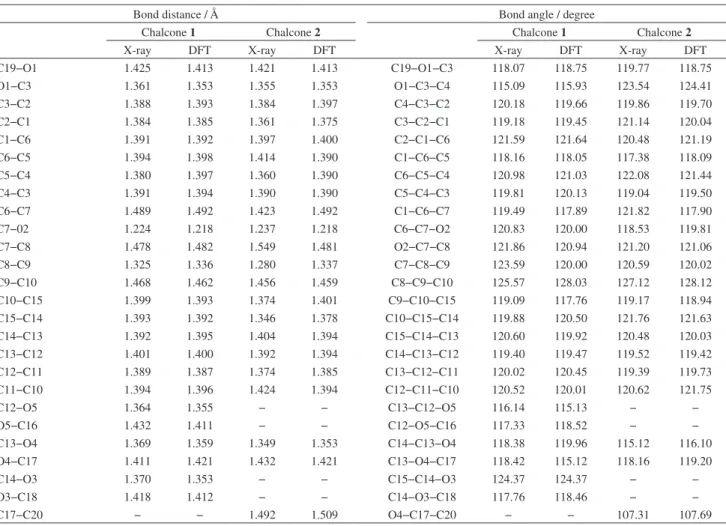
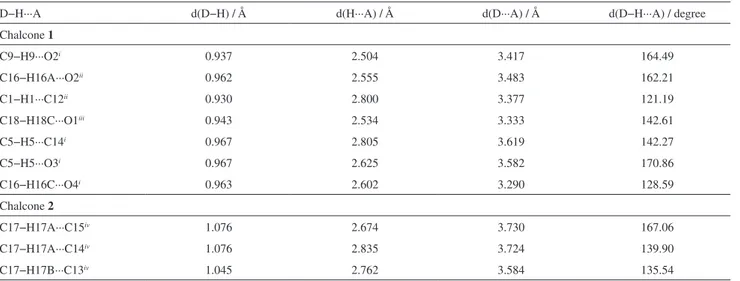
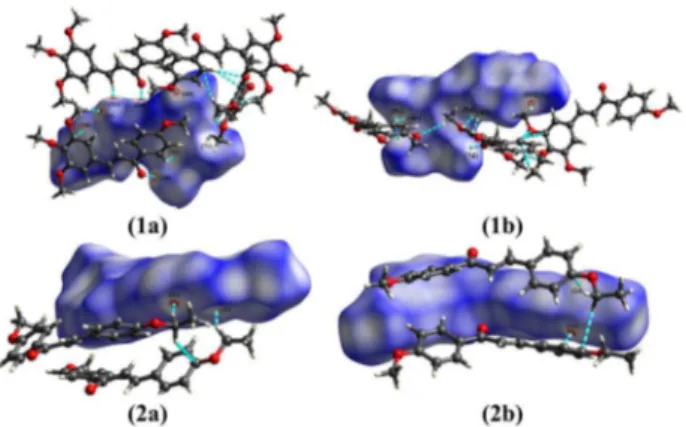
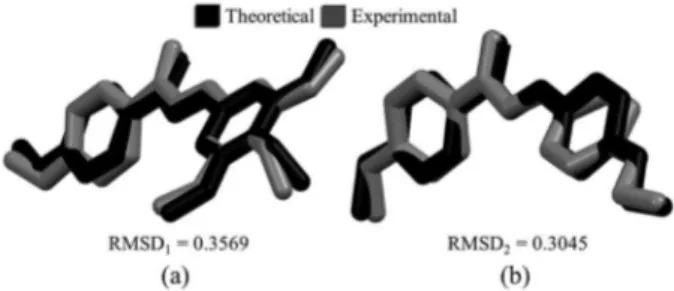
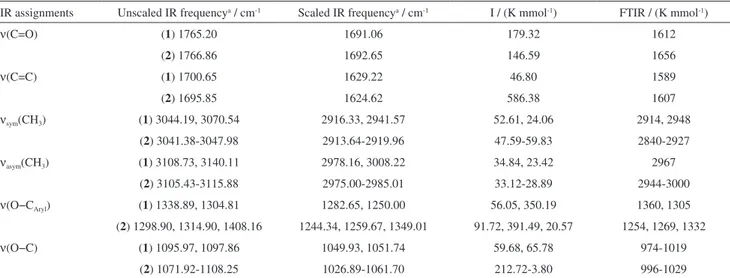
Documentos relacionados
After the construction of 2D and 3D QSAR models, we performed theoretical calculations of some molecular properties, such as the maps of molecular orbitals (highest
Em geral, as propriedades são largamente determinadas pelas energias dos orbitais de fronteira : o orbital molecular ocupado de maior energia ( highest occupied
In the present study, the thirty screened out phytochemicals were evaluated based upon DFT analysis and the lower band energy gap of molecular orbital energies
method, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies were calculated to characterize global chemical reactivity
The probability of attending school four our group of interest in this region increased by 6.5 percentage points after the expansion of the Bolsa Família program in 2007 and
Using a deformed exponential function and the molecular-orbital theory for the simplest molecular ion, two new analytical functions are proposed to represent the potential energy
When a molecule absorbs light in the UV-Vis region, an electron is promoted from the highest-energy occupied molecular orbital (HOMO) to the lowest- energy unoccupied
In the Raman spectrum, the intensity of the bands attributed to the hydroxyl units are far more intense than the bands assigned to water stretching vibrations. This is be- cause
