Ce travail a été réalisé au sein du Laboratoire de Chimie Organique et Bioorganique de l'Ecole Nationale Supérieure de Chimie de Mulhouse. Je remercie le Professeur Claude Le Drian de m'avoir accueilli dans son laboratoire et je lui demande de trouver ici un témoignage de ma gratitude pour l'intérêt qu'il porte à ces travaux.
Introduction
Les glycosides
- Diversité des glycosides
- Les cyanoglucosides cyanogènes
- Les cyanoglucosides non-cyanogènes
- Structures et propriétés chimiques
- Occurrence naturelle et propriétés biologiques
- Conclusion
Lorsque ces plantes de la famille des Crucifères (nom actuel : Brassicaceae) sont attaquées, une enzyme coupe la partie sucre et libère de l'isothiocyanate d'allyle (responsable du goût piquant de la moutarde) qui repousse l'animal attaquant. La ménisdaurine a été isolée en 19786) à partir de Menispermum dauricum (162 mg par kg de plante coupée), plante médicinale de la famille des Menispermacées.
La réaction de glycosidation
- Généralités
- Exemples
- Glycosidation du Ménisdaurilide
- Hémisynthèse de la Simmondsine
- La réaction de Koenigs-Knorr optimisée au laboratoire
- Cas de la Bauhinine
- Cas du Lithospermoside
- Cas du cyanoglucoside isolé de l’Ilex warburgii
- Conclusion
Schéma 36 : Résultats de glycosidation en fonction de la nature du groupe protecteur dans l'aglycone C(4). Schéma 38 : Résultats de glycosidation selon la nature du groupe protecteur dans l'aglycone C(4) et C(5).
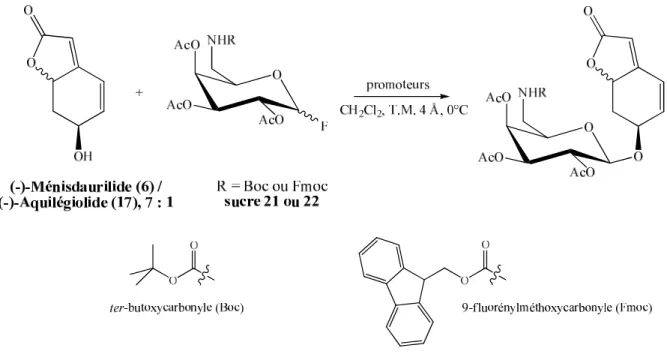
Synthèse d’aglycones de type cyclitol
- Hémisynthèse de la Simmondsine et de la Purshianine
- Synthèse du Ménisdaurilide
- Synthèse à partir de la chimie des « sucres nus »
- Généralités
- Synthèses antérieures effectuées sans ouverture d’époxyde
- Synthèses antérieures effectuées avec ouverture d’époxyde
- Synthèse du muco-quercitol, du D-chiro-inositol et de l’allo-inositol
- A partir de la synthèse du Ménisdaurilide et de dérivés proches
- A partir de la chimie des « sucres nus »
- Conclusion
L'efficacité de la réaction de glycosidation est essentielle, mais la préparation de l'aglycone représente encore une part importante du travail. Le groupe hydroxyle sur α de la cétone est protégé par un éther TBDMS et la réaction d'oléfination de Peterson assure la formation préférentielle de l'isomère Z. Après l'étape de formation de la double liaison α,β-insaturée et le clivage de l'éther TBDMS, un mélange A inséparable d'Aquilegiolide et de Menisdaurilide est obtenu.
Après avoir inversé la configuration du carbone 6 avec DBU, Ueda et son groupe ont rencontré des problèmes lors de l'étape de coupure du groupe protecteur MOM de OH à C(6). En 2007, Ueda et son groupe ont publié une synthèse complètement différente du (-)-ménisdaurilide avec le 1,4-cyclohexadiène comme matière première,84) similaire à la synthèse du (±)-ménisdaurilide par Mori et son groupe43) (voir pp. 47-48). La synthèse commence par la saponification de l'ester (+) -89, la formation d'acétal de benzyle (selon les conditions de Noyori) de l'oxanorbornénone (+) -88 résultante et l'époxydation de la double liaison jusqu'à l'utilisation de l'acide métachloroperbenzoïque. . a), b), c). e), f). h).
Selon les conditions de réduction de la fonction cétone de cet intermédiaire (-)-106, on obtient soit le composé (-)-107, soit son épimère (-)-108 (respectivement à l'aide du DIBAH, ou du réactif de Luche). . Josien-Lefebvre a mis à profit l'expérience acquise lors de la synthèse du (-)-Bauhinin, pour réaliser efficacement la synthèse du (-)-Lithospermoside en 2003.53. La bis-hydroxylation de la double liaison de l'intermédiaire (+)-121 et l'hydrogénolyse catalytique du cycle dioxane permettent d'obtenir le muco-quercitol en 7 étapes et un rendement global de 9%.
Synthèse totale de la (-)-Ménisdaurine
Synthèse de l’aglycone de la Ménisdaurine
- Première voie de synthèse : une étape de SN 2
- Exemples de SN 2 réalisée sur des dérivés de « sucres nus »
- Préparation d’halogénocétones, produits de départ de la SN 2
- Essais de SN 2 réalisée sur les halogénocétones
- Préparation d’hydroxycétones
- Deuxième voie de synthèse : une étape d’ouverture d’époxyde
- Ouverture de l’époxyde par un dérivé sélénié
- Ouverture de l’époxyde par un dérivé soufré
- Récapitulation
- Etapes de fin de synthèse de l’aglycone de la Ménisdaurine
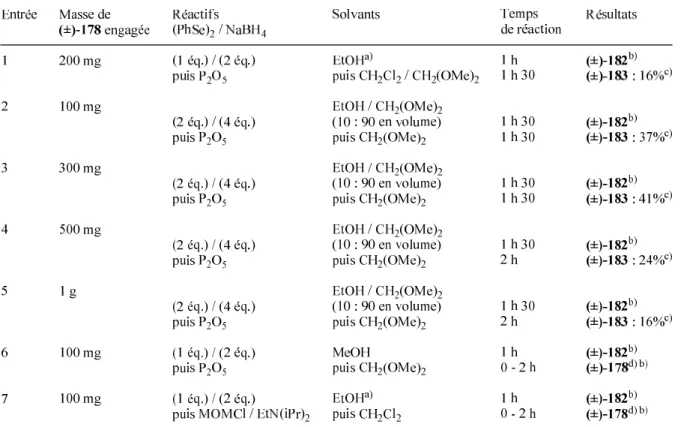
Schéma 79 : Ouverture de l'époxyde du composé (±)-179 dans des conditions nouvelles (I2, acétone) et protection par un groupe MOM de l'intermédiaire obtenu (±)-227. L'absence d'acétone dans le milieu réactionnel doit donc empêcher la formation d'époxy cétone (±)-178 et favoriser la formation du produit (±)-227 issu de l'ouverture de l'époxy. En effet, lors d'un test réalisé dans le diméthoxyméthane, aucune trace d'époxy cétone (±)-178 n'a été détectée, seuls les produits issus de l'ouverture de l'époxy ont été obtenus (mais non isolés).
L'ouverture de l'époxy est facilement réalisée dans ces conditions douces, mais le produit (±)-227 n'est pas le produit principal de la réaction. Comme dans le cas de l'ouverture de l'époxyde (±)-178 dans les conditions de Sharpless124) ((PhSe)2 et NaBH4), la réaction dans ces nouvelles conditions est complètement stéréospécifique (OH en C(5) en position exo ) et complètement régiosélectif (groupe phénylthio ou atome d'iode en C(6)). En revanche, l'intermédiaire (±)-230 a pu être isolé avec un rendement proche (70%) du rendement maximum attendu (75%) en déposant directement l'huile brute réactionnelle issue de l'étape d'ouverture de l'époxy (±) -178 sur colonne de silice.
Les auteurs ont alors émis l'hypothèse qu'un acétal d'oxanorbornénone (±)- 88 doit subir un réarrangement similaire en présence de dibrome : le composé (-)- 243 est traité par la dibromine dans le dichlorométhane à −90°C et conduit au composé (+ )- 205 par migration intramoléculaire 1,3-endo d'un groupe benzyloxy. Il est également important de noter que seule la formation de produits finaux résultant de l’ouverture de l’époxyde par attaque C(6) est observée. Il est possible que l'ouverture de l'époxyde d'époxycétone (±)-178 dans ces conditions soit réalisée par un mécanisme faisant également intervenir principalement une attaque du côté du site endo.
Schéma 96 : Mécanisme d'ouverture de l'époxyde du composé (±)-178 par ajout de PhSH en présence de I2 ou IBr. Schéma 99 : Coupure de l'acétal de méthyle du composé (±)-144 et protection de l'OH libre par un groupe MOM.
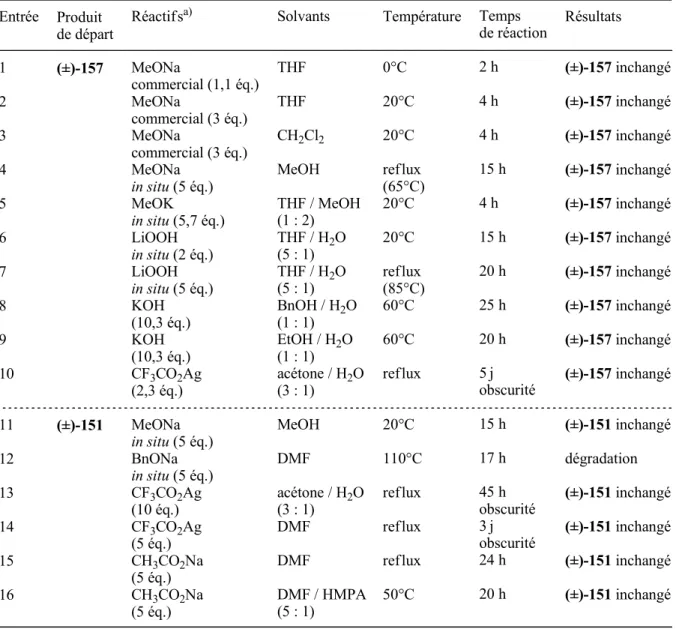
Etape de glycosidation
La ménisdaurine aglycone (±)- 278 a finalement pu être préparée avec un rendement global satisfaisant de 24 % en 8 étapes à partir du cyanoacétate (±)- 90 . Des conditions douces de glycosidation selon la méthode Koenigs-Knorr développée en laboratoire (voir section 2.3., chapitre 1) ont été appliquées à l'aglycone Menisdaurine (±)-278. Plusieurs tests de glycosidation de l'aglycone protégée de Menisdaurine (±) -278 sont rapportés dans le tableau 11 (voir p. 130).
En revanche, dans un environnement fortement basique, c'est-à-dire lorsque la totalité de la base à ajouter (2 éq.) est présente dès le début de la réaction, la formation des deux orthoesters 281 et/ou 282 est la réaction dominante. Par conséquent, la formation des différents produits de glycosidation de l'aglycone (±)-278 de Menisdaurin apparaît extrêmement sensible à la quantité de base présente au début de la réaction, encore plus que celle observée pour l'aglycone (±)-41 protégée cyanoglucoside. isolé de Ilex warburgii (voir p. 43, partie 2.3.3., chapitre 1). Toute la difficulté de cette étape de glycosidation réside dans le compromis qu'il faut trouver entre limiter la formation prédominante d'orthoesters 281 et/ou 282 en milieu basique, limiter la formation de produits secondaires issus de la coupure du groupe protecteur MOM en milieu basique. environnement. acide, et la formation avec un rendement satisfaisant du β-D-glucoside 279.
De même, plus la quantité de base ajoutée au début de la réaction est faible, plus le rendement en orthoesters diminue et plus le rendement en produits secondaires résultant de la coupure du groupe protecteur MOM augmente. Le rendement en β-D-glucoside 279 n'était que de 13 % tandis que les produits secondaires résultant du clivage du groupe protecteur MOM ont été obtenus avec un rendement global de 35 %. Dans ces conditions, aucune formation de produits secondaires n'a été observée suite au clivage du groupe protecteur MOM.

Fin de la synthèse
Nous avons donc tenté de réaliser l'isomérisation de la double liaison après la coupure du groupe MOM dans l'espoir que la séparation des composés C(4) déprotégés 286(E) et 286'(Z) serait plus facile. L'isomérisation de la double liaison du composé 286(E) a été réalisée dans les mêmes conditions réactionnelles que lors des tests précédents réalisés avec le β-D-glucoside 279(E). Cependant, l’étape d’isomérisation des doubles liaisons fonctionne plus efficacement lorsqu’elle est réalisée à partir du β-D-glucoside 279(E).
Concernant l'étape d'isomérisation des doubles liaisons (étape a) ou c)), des problèmes de reproductibilité ont été rencontrés quel que soit le composé de départ utilisé (279(E) ou 286(E)). Ainsi, la synthèse totale du pentaacétate de ménisdaurine (287) a été réalisée en 12 étapes à partir de cyanoacétate (±) -90 et un rendement global de 2 %. L'analyse rétrosynthétique rapide de l'aglycone Menisdaurin indique clairement que la préparation d'un composé C est l'étape clé.
Aucun problème majeur n'a été rencontré lors des dernières étapes de synthèse de l'aglycon protégé de la Ménisdaurine (±)-278 qui a finalement pu être préparé avec un rendement global de 24% en 8 étapes à partir du cyanoacétate (±)-90. Des difficultés de reproductibilité de nos tests lors de l'étape d'isomérisation de la double liaison portant le groupement nitrile ont été rencontrées. La synthèse totale du pentaacétate de ménisdaurine (287) a été réalisée en 12 étapes à partir de cyanoacétate (±) -90 et un rendement global de 2 %.
Généralités
- Solvants, réactifs et réactions
- Chromatographie sur couche mince
- Chromatographie sur colonne
- Point de fusion et pouvoir rotatoire
- Spectre UV et spectre de masse
- Spectre de résonance magnétique nucléaire
- Description des spectres
- Analyse élémentaire
Les spectres de résonance magnétique nucléaire du proton (RMN 1H) et du carbone (RMN 13C) ont été enregistrés à 298 K, sur un instrument Bruker Advance 400 (400 MHz pour la RMN du proton et 100,58 MHz pour la RMN du carbone). Les déplacements chimiques δ des protons sont exprimés en ppm, les références sont 7,26 ppm pour CDCl3, 2,50 ppm pour DMSO-d6 et 3,30 ppm pour CD3OD. Les déplacements chimiques δ des carbones sont exprimés en ppm, la calibration est effectuée sur le pic central du triplet à 77,00 ppm pour CDCl3.
Les attributs subtils des signaux RMN 1H et RMN 13C de plusieurs composés ont été résolus grâce à l'analyse des spectres de corrélation 2D 1H-1H (CoSy) et 1H-13C (HSQC et HMBC). Les abréviations suivantes sont utilisées pour décrire la pluralité de signaux : s (simple), d (double), t (triple), quadruple. Les analyses élémentaires (EA) ont été réalisées par le Service Central d'Analyses du CNRS, au Département d'Analyses Élémentaires de Vernaison.
Synthèse totale de la (-)-Ménisdaurine
Le suivi de la réaction par CCM indique une disparition totale du produit de départ (±)-167 après 1 heure et 15 minutes de réaction. La phase organique est ensuite séchée sur MgSO 4 , filtrée, concentrée sous pression réduite et le résidu obtenu est purifié par chromatographie sur colonne de gel de silice (éluant : AcOEt/EP, 1:2). Le suivi de la réaction par CCM indique une disparition totale du composé intermédiaire (±)-182 (après 1 heure et 45 minutes de réaction) et l'apparition d'un nouveau composé (composé (±)-183).
Le milieu réactionnel est concentré sous pression réduite et 70 mL de solution aqueuse saturée de NaHCO3 sont ajoutés. Le suivi de la réaction par CCM montre la disparition complète de l'intermédiaire (±)-227 (après 50 min de réaction) et l'apparition d'un nouveau composé (composé (±)-228). Le milieu réactionnel est concentré sous pression réduite et 50 mL de solution aqueuse saturée de Na2CO3 sont ajoutés.
La réaction est suivie par RMN 1H (CDCl3) des résidus obtenus après concentration des échantillons de la matière première réactionnelle sous pression réduite. Le déroulement de la réaction est suivi par RMN 1H (CDCl3) des résidus obtenus ultérieurement. A la fin de l'addition lente, la solution réactionnelle est agitée pendant 16 heures à température ambiante, sous argon et à l'abri de la lumière.
En fin de réaction, le brut réactionnel est filtré sur Célite, rincé à l'AcOEt et le filtrat est concentré sous pression réduite. Le suivi de la réaction par CCM indique la disparition quasi totale du produit de départ 279(E) après 2 heures de réaction.