Synthèse asymétrique de l'épi-jasmonate de méthyle et de son énantiomère (ent-épi-jasmonate de méthyle) par voie chimique et enzymatique. Les prostaglandines, les prostacyclines et les thromboxanes sont biosynthétisées à partir de l'acide arachidonique chez les mammifères [8,9,10].
Origines, diversité, propriétés
- Structure chimique et stéréochimie
- Les épimères du jasmonate de méthyle
- Les oxylipines au sein des plantes
- Les jasmonoïdes et les oxylipines dans le milieu naturel
- Les jasmonoïdes et les oxylipines dans le milieu terrestre
- Les jasmonoïdes et les oxylipines dans le milieu marin
Isolés pour la première fois en 1962 en tant que composant essentiel de l'huile de fleur de jasmin, Jasminum grandiflorum L., les stéréoisomères du jasmonate de méthyle sont des composés de signalisation omniprésents dans les plantes. Il s'agit du premier rapport indiquant que les jasmonates de méthyle ont un effet régulateur sur la croissance des plantes.

Biosynthèse des jasmonoïdes et oxylipines
- Biosynthèse des cis-jasmonates de méthyle et des oxylipines végétales
- Degré d’épimérisation de l’épi-jasmonate de méthyle au sein des plantes
- Biosynthèse enzymatique des dérivés amino-conjugués de l’acide épi-jasmonique
- Autres jasmonoïdes biosynthétiques
Ils sont biosynthétisés à partir de l'acide épijasmonique par l'action de deux enzymes : Jasmonate Résistant 1 ou 4 (JAR1 ou 4) [57]. Schéma 12 : Séquence biosynthétique des principaux dérivés aminés conjugués de l'acide épi-jasmonique avec JAR1 et 4.
Propriétés et fonctions des jasmonates de méthyle
- Propriétés organoleptiques des jasmonates : applications en parfumerie
- Seuils de détection olfactive des stéréoisomères du jasmonate de méthyle
- Synthèses totales de la cis-Hédione ® et de la Paradisone ®
- Rôle chez les végétaux
- Croissance et développement
- Réponse à des facteurs environnementaux
Régulation de la croissance et du développement mais aussi induction des mécanismes de défense (Schéma 22). Schéma 22 : Rôle omniprésent des jasmonates de méthyle et des oxylipines : contrôle de la croissance et du développement et protection contre les stress biotiques et abiotiques.

3.2.2.1. Réponse à des facteurs biotiques
Enfin, lorsqu’elle est attaquée par un insecte herbivore, la plante synthétise des molécules volatiles, dont le jasmonate de méthyle et le cis-jasmone, qui sont ensuite libérées. Ces phéromones volatiles attirent d'autres insectes qui sont susceptibles de parasiter l'insecte herbivore à l'origine de l'attaque et de le tuer à court terme.
3.2.2.2. Réponse à des facteurs abiotiques
- Relation structure-propriétés des jasmonates de méthyle au sein des plantes
- Rôle chez les algues
- Elicitation de métabolites secondaires grâce aux jasmonates de méthyle
- Elicitation des organismes photosynthétiques marins
- Elicitation des organismes photosynthétiques terrestres
- Conclusion
Schéma 24 : Modulations chimiques et stéréochimiques du jasmonate de méthyle : relation structure/activité chez les plantes. Schéma 26 : Induction de la biosynthèse de molécules toxiques dans les plantes par le jasmonate de méthyle.
![Figure 5 : Perception et régulation des gènes jasmonate-dépendants par la N-(+)-7-épi-jasmonoyl-L- N-(+)-7-épi-jasmonoyl-L-isoleucine, les protéines JAZ, et le complexe ubiquitine-ligase [108] .](https://thumb-eu.123doks.com/thumbv2/1bibliocom/465302.70576/45.892.119.765.456.842/perception-régulation-jasmonate-dépendants-jasmonoyl-isoleucine-protéines-ubiquitine.webp)
LES JASMONATES DE MÉTHYLE : NOUVEAUX AGENTS
- Introduction : le cancer
- La voie PI-3-K/Akt
- Voie de signalisation des PI-3-K/Akt pour le développement cellulaire
- Voie de signalisation des PI-3-K/Akt pour l’apoptose
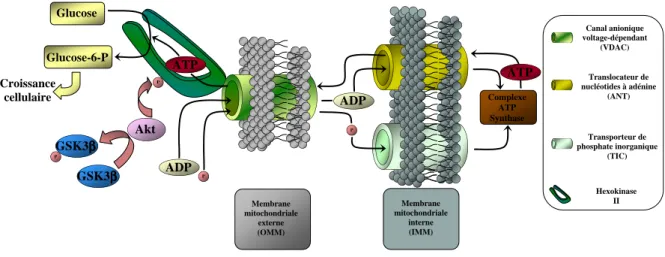
- L’hexokinase II : cible mitochondriale du jasmonate de méthyle
- Autres molécules ciblant les canaux mitochondriaux
- Activités anticancéreuses des jasmonates de méthyle
- Cytotoxicité sur les cellules du neuroblastome humain
- Effets anticancéreux sur les cellules cancéreuses du sein et de la prostate
- Effets cytotoxiques sur les cellules du carcinome cervical
- Effets sur les cellules cancéreuses du poumon à non petites cellules
- Effet du jasmonate de méthyle sur le parasite amitochondrial T. Vaginalis
- Effets synergétiques et cytotoxicité coopérative
- Optimisation de la cytotoxicité des jasmonates de méthyle
- Conclusion
Acide dichloroacétique : Il modifie le métabolisme de la glycolyse à la phosphorylation oxydative dans les cellules cancéreuses [179]. Au cours de la dernière décennie, les jasmonates de méthyle sont devenus de bons candidats pour de nouveaux traitements contre le cancer.
![Figure 6 : Voies de signalisations des PI-3-K pour le développement cellulaire [136]](https://thumb-eu.123doks.com/thumbv2/1bibliocom/465302.70576/54.892.168.703.106.426/figure-voies-signalisations-pi-k-développement-cellulaire-136.webp)
RAPPELS SUR LES SYNTHÈSES TOTALES DES JASMONATES DE
Caractéristiques communes des différentes synthèses
Schéma 29 : Principales méthodes utilisées pour la préparation des fonctions ester de cyclopentanone, d'oléfine et de méthyljasmonate. La plupart des équipes qui ont initié la synthèse des jasmonates de méthyle ont des stratégies basées essentiellement sur la même logique de synthèse pour la préparation de chaînes α et ω fonctionnalisées. Historiquement, les premières synthèses mises en œuvre concernaient la synthèse du (±)-jasmonate de méthyle racémique.
Ce n’est qu’à partir du début des années 1990 que les synthèses totales de méthyl-épi-jasmonate et d’ent-épi-jasmonate énantiopurs ont été envisagées.
- Synthèses du (±)-jasmonate de méthyle racémique
- Sisido et al. – 1968: préparation d’une oléfine bicyclique fonctionnalisée
- Büchi et al. – 1971 : fonctionnalisation d’une cyclopentanone en α puis β
- Decorzant et al. – 1978. Tsuji et al. – 1987: fonctionnalisation d’une cyclopentanone en α puis β
- Negishi et al. – 1985 : β - puis α -fonctionnalisation d’une cyclopenténone
- De Saint Laumer et al. – 2006 : préparation d’une chloroindénone par Diels-Alder
- Synthèse des ( − )-jasmonate et (+)-jasmonate de méthyle énantiopurs
- Quinkert et al. – 1982 : préparation d’une cyclopenténone α , β -disubstituée
- Posner et al. – 1985 : β -fonctionnalisation d’une cyclopenténone α -substituée
Schéma 31 : Synthèse du (±)-méthyljasmonate et du cis-jasmone selon Büchi et al. La condensation du malonate de méthyle sur la diénone cyclique obtenue suivie d'une décarboxylation à chaud donne alors du (±)-jasmonate de méthyle avec un rendement global de 31 %. Avec cette nouvelle stratégie, le (±)-méthyljasmonate est obtenu avec un rendement global de 60% en quatre étapes.
Enfin, une réaction de Wittig permet d'obtenir le (±)-jasmonate de méthyle avec un rendement global d'environ 42 %.
Synthèses totales des épi-jasmonate et ent-épi-jasmonate de méthyle
- Synthèses du (±)-cis-jasmonate de méthyle racémique
- Sarkar et al. – 1997 : cyclisation d’un diène activé par thermolyse
- Hailes et al. – 2001 : préparation d’une 5-méthylindén-1-one par réaction de Diels-Alder ionique
- Synthèses totales des épi-jasmonate et ent-épi-jasmonate de méthyle énantiopurs
- Helmchen et al. – 1990 : préparation d’un jasmonoïde clé, la δ -jasmolactone
- Kitahara et al. – 1991 : préparation d’un jasmonoïde clé, la δ -jasmolactone
- Fehr et Galindo – 2000 : Réarrangement de Claisen d’un cétène d’acétal silylé
- Inomata et al. – 2004 : synthèse d’un précurseur bicyclique par RDAE
- Taapken et al. – 1994 : préparation d’une indénone fonctionnalisée
Une oxydation finale donne ensuite du méthyl-(±)-cis-jasmonate avec un rendement total de 8 %. Il est structurellement proche du matériau utilisé par Torii pour la synthèse de l'épi-jasmonate de méthyle [218]. Schéma 44 : Synthèse d'épi-jasmonate de méthyle bloqué stéréochimiquement en position C7 par un groupe méthyle selon Taapken et al.
L'épi-jasmonate méthylé en C7 est obtenu après réaction de Wittig, puis oxydation de l'alcool libre avec un rendement global de 32 %.
Conclusion : Récapitulatif et comparaison des différentes synthèses
Pour synthétiser du méthyl-cis-jasmonate énantiopur, nous avons souhaité utiliser des précurseurs bicycliques énantiopurs de type diéno- ou diexo-cis-bicyclo[3.3.0]octane-2,6-diol de symétrie C2. La jonction cis-cyclique de ces diols bicycliques énantiopurs définit alors la stéréochimie du jasmonate cis-méthylique ciblé. Ainsi, l'épi-jasmonate de méthyle sera alors obtenu à partir de diols dont la jonction cyclique est de stéréochimie (S,S).
Schéma 45 : Schémas rétrosynthétiques du méthyl-épi-jasmonate et de l'ent-épi-jasmonate à partir de diols énantiopurs de symétrie C2, de sorte que la liaison cyclique soit cis.
Les catalyseurs de Jacobsen : structures, applications, réactivités et synthèses
- Structures et modulations des catalyseurs de Jacobsen
- Méthodes générales de synthèse des catalyseurs de Jacobsen
- Préparation des (R,R)- et (S,S)(Salen)Co(III)X
- Préparation des (R,R)- et (S,S)(Salen)M(III)X (M = Cr, Mn)
- Préparation des (R,R)- et [(S,S)(Salen)M(V)=O]X (M = Cr, Mn)
- Préparation d’autres catalyseurs métalliques
- Additions nucléophiles sur les époxydes
- Résolution cinétique hydrolytique d’époxydes racémiques (HKR)
- Réactivité des méso-époxydes
- Autres réactions utilisant les catalyseurs de Jacobsen
État d'oxydation +V : l'atome métallique porte un substituant Les bases chirales de Schiff sont couramment utilisées pour cette dernière réaction, la résolution cinétique d'un époxyde racémique ou la désymétrisation de mésoépoxydes achiraux. Selon la nature de l'époxyde, les catalyseurs de Jacobsen permettent deux principaux types de réactions : résolution cinétique hydrolytique (HKR : « Hydrolytic Kinetic Résolution ») de l'époxyde final ou ouverture asymétrique des méso-époxydes achiraux.
Cette réaction a lieu en présence d'un catalyseur chiral fonctionnalisé avec un atome central de cobalt à l'état d'oxydation (+III) et un contre-ion, généralement de l'acétate (Schéma 58).

Préparation des catalyseurs de Jacobsen : Métallation et fonctionnalisation de ligands tétradentates…
- Synthèse des (R,R)- et (S,S)(Salen)Co(II)
- Synthèse des (R,R)- et (S,S)(Salen)M(III)X
- Synthèse des (R,R)- et (S,S)(Salen)M(III)Cl
- Synthèse des (R,R)- et (S,S)(Salen)Co(III)OH
Fonctionnalisation de bases de Schiff sous la forme (R,R)(Salen)Co(III)OH en présence de sels d'hydroxyde. La même unité catalytique catalyserait à la fois l'ouverture de l'époxyde et la cyclisation assistée par électrons du méso-époxyde. Un homologue du motif catalytique B (noté C) de type (R,R) ou (S,S)(Salen)Co(IV)X-OH, se formerait alors in situ en présence d'eau (Schéma 67).
Schéma 67 : Nouvelle stratégie pour la cyclisation assistée par électrons du méso-époxyde, une unité catalytique (R, R)(Salen)Co(IV)X-OH formée in situ (un énantiomère illustré).

Réactivité du méso-époxyde dérivé du cycloocta-1,5-diène
- Synthèse du (Z)-(1S,8R)-9-oxa-bicyclo[6.1.0]non-4-ène
- Essais de la réaction de cyclisation assistée par les catalyseurs
- Effet du contre-ion « X »
- Etat de transition
En revanche, nous avons pu mettre en évidence l'hydrolyse énantiosélective du méso-époxyde en diol homochiral 10A ou 10B, selon la stéréochimie du catalyseur utilisé. Les résultats obtenus pour les rendements et les énantiosélectivités de l'hydrolyse des méso-époxydes en fonction de la charge catalytique sont résumés dans le tableau suivant (Tableau 8). Seuls les produits d'hydrolyse énantiosélective du méso-époxyde en diol homochiral 10A et 10B ont été observés (Tableau 9).
Schéma 71 : État de transition possible lors de l'hydrolyse énantiosélective du mésoépoxyde en diol homochiral (R,R) ou (S,S) en présence de catalyseur (S,S)SalenCo(III)X ou

Méthodes générales
- Conduite des réactions
- Purification des solvants et des réactifs
Les sels hautement hygroscopiques tels que le triflate de scandium (III), l'acétate de cobalt (II), le chlorure de chrome (II) et le chlorure de lithium ont été systématiquement séchés sous vide à l'aide d'une pompe à vide pendant 24 à 48 heures avant utilisation.
Analyses
- Spectroscopie de résonance magnétique nucléaire
- spectroscopie d’absorption infrarouge
- Polarimétrie
- Point de fusion
- Spectroscopie de masse haute résolution
- Analyse en impact électronique (EI)
- Analyse en électronébulisation (ESI)
La rotation de la lumière polarisée par les produits dissous dans le chloroforme a été mesurée dans une cuve de 10 cm de long à une température constante de 20°C. Les points de fusion, exprimés en degrés Celsius (°C), sont mesurés sur banc Kofler et ne sont pas corrigés. Les analyses d'impact électronique ont été effectuées sur un spectromètre de masse VARIAN MAT 311 à double focalisation haute résolution (géométrie NIER-JOHNSON BE inversée).
La précision obtenue en mesurant la masse exacte des ions est de 4 chiffres significatifs (attribution à la formule brute).
6.5.2.1. Micromass ZABSpecoaTOF
6.5.2.2. Bruker MicrO-TOF Q I
La détermination des masses précises est effectuée par correspondance de pics en utilisant du perfluorokérosène (PFK) comme référence interne.
6.5.2.3. Waters Q-TOF 2
- Détermination des excès énantiomériques par dosage HPLC
- Modes opératoires généraux
- Dosage d’une solution commerciale de n-butyllithium
- Préparation du diisopropylamidure de lithium (LDA) et de l’hexaméthyldisilylamidure de lithium
- Préparation de la résine DOWEX OH -
- Réactions de coupure ozonolytique
- Réactions sous champ micro-onde
- Résolutions enzymatiques
Les phases organiques combinées ont été lavées avec une solution saturée d'hydrogénocarbonate de sodium (200 ml), séchées sur sulfate de magnésium, filtrées et concentrées sous vide partiel. Les phases organiques combinées ont été lavées avec une solution saturée de bicarbonate de sodium (150 ml), séchées sur sulfate de magnésium, filtrées et concentrées sous vide partiel. Les phases organiques combinées ont été lavées avec une solution saturée d'hydrogénocarbonate de sodium (40 ml), puis avec de la saumure (35 ml), séchées sur sulfate de magnésium et concentrées sous vide partiel.
Les phases organiques réunies sont lavées avec une solution saturée d'hydrogénocarbonate de sodium (20 mL) puis de la saumure (20 mL), séchées sur sulfate de magnésium et concentrées sous vide partiel.
SYNTHÈSE DE PRÉCURSEURS CHIRAUX BICYCLIQUES PAR VOIE
Synthèse de diols cycliques homochiraux énantiopurs par réaction enzymatique
- Synthèse de précurseurs racémiques homochiraux
- Résolution enzymatique du (Z)-(1R*,2R*)-cyclooct-5-ène-1,2-diol ou de l’acétate de (Z)-(1R*,8R*)-8-
Nous avons tout d'abord considéré la résolution enzymatique du diol 10 racémique en milieu organique par transestérification de l'acétate de vinyle en présence de lipase de Pseudomonas cepacia capable d'agir dans le THF à une température de 55°C (Figure 76). Schéma 77 : Test de dissolution du diol homochiral (Z)-(1R*,2R*)-cyclooct-5-ène-1,2-diol par transestérification de l'acétate de vinyle en présence de lipase de Pseudomonas cepacia. La réaction de transestérification de l'acétate de vinyle avec le mélange racémique des diols 10A et 10B permet de synthétiser l'hydroxyester 12 correspondant.
Schéma 78 : Résolution enzymatique en milieu tamponné d'acétate racémique de (Z)-(1R*,8R*)-8-acétoxy-cyclooct-4-ényle en présence de lipase de Pseudomonas cepacia.

Monoestérification des (Z)-(1S,2S)- et (Z)-(1R,2R)-cyclooct-5-ène-1,2-diols énantiopurs
- O-monoacétylation du (Z)-(1S,2S)-cyclooct-5-ène-1,2-diol
- O-monobenzoylation des (Z)-(1S,2S)- et (Z)-(1R,2R)-cyclooct-5-ène-1,2-diols
La O-monobenzoylation à 0 °C dans des conditions alcalines des diols 10A et 10B dans le dichlorométhane par addition de bromure de benzoyle est réalisée avec d'excellents rendements de 97 % et 91 %, respectivement (schéma 83). Nous disposons désormais de quatre hydroxyesters énantiopurs, deux dans les séries O-acétate 12A et 12B et deux dans les séries O-benzoate 13A et 13B. La protection permettra de préparer les synthons appropriés qui permettront de synthétiser les précurseurs des diols bicycliques énantiopurs.

Protection des (Z)-(1S,2S)- et (Z)-(1R,2R)-cyclooct-5-ène-1,2-diols O-monoacétylés et O-
- Stratégie alkyle : alkylation des (Z)-(1S,2S)- et (Z)-(1R,2R)-cyclooct-5-ène-1,2-diols O-monoacétylés…
- Stratégie silyle : silylation du (Z)-(1S,2S)- et (Z)-(1R,2R)-cyclooct-5-ène-1,2-diols O-monoacétylés et
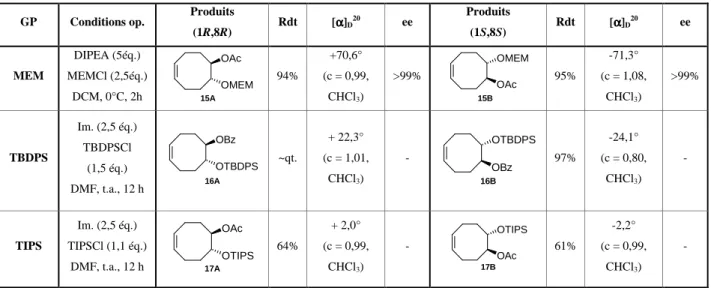
Pour moduler le groupe protecteur de la fonction alcool, nous avons réalisé les silylations des diols énantiopurs O-monoacétylés 12A et 12B et O-monobenzoylés 13A et 13B dans du DMF, à température ambiante, en présence d'imidazole, et d'un alkyl chlorosilane (Schéma 86). Les rendements observés pour la silylation des hydroxyesters O-benzoylés en 13A et 13B sont quasiment quantitatifs, ce qui n'est pas le cas pour les dérivés O-acétylés 12A et 12B. En utilisant de la 2,6 lutidine (3 équiv.) et du TIPSOTf (2,5 équiv.) dans du dichlorométhane à 0°C, la silylation de l'hydroxyester 12A O-acétylé est réalisée avec un rendement moyen de 53 %.
Un résumé des esters fonctionnalisés avec leurs groupes protecteurs respectifs est présenté dans le tableau suivant (Tableau 12).
Hydrolyse des esters de (Z)-8-hydroxy-cyclooct-4-ényles énantiopurs
- Hydrolyse des acétates de (Z)-(1S,2S)- et (Z)-(1R,2R)-8-hydroxy-cyclooct-4-ényles O-alkylés et O-
- Réduction des benzoates de (Z)-(1S,2S)- et (Z)-(1R,2R)-8-hydroxy-cyclooct-4-ényles O-silylés
La déprotection des (Z)-8-hydroxy-cyclooct-4-énylbenzoates O-silylés 16A et 16B a été réalisée dans du THF anhydre à basse température en présence de deux équivalents d'hydrure de diisobutylaluminium [260] . Schéma 90 : Réduction des benzoates O-silylés de (Z)-(1S,2S)- et (Z)-(1R,2R)-8-hydroxy-cyclooct-4-ényl en présence d'hydrure de diisobutylaluminium. Les deux alcoxyalcools cycliques insaturés 18A et 18B et les silanyloxyalcools 19A et 19B peuvent maintenant être fonctionnalisés avec un bon groupe partant afin de synthétiser ensuite les dérivés bicycliques de SN2 (Schéma 90).

Méthanesulfonation
Les alcools sulfonylés 20A à 21B sont maintenant fonctionnalisés de telle sorte que le groupe partant mésyle approprié pourrait permettre une cyclisation intramoléculaire par substitution nucléophile assistée par double liaison. Lors de cette cyclisation, la fonction alcool manquante pourrait être assurée par l'eau, qui jouerait en partie le rôle de solvant. Par la suite, cette réaction de cyclisation peut être optimisée en utilisant d'autres systèmes de solvants polaires.
Le groupe méthyle pourrait être remplacé par un groupe tosyle ou du collagène pour améliorer le rendement.
Conclusion
Les phases organiques réunies sont lavées avec une solution saturée d'hydrogénocarbonate de sodium (50 mL), puis avec de la saumure (50 mL), séchées sur sulfate de magnésium, filtrées et concentrées sous vide partiel. Les phases organiques combinées sont lavées avec une solution saturée d'hydrogénocarbonate de sodium (30 mL), puis avec de la saumure (30 mL), séchées sur sulfate de magnésium, filtrées et concentrées sous vide partiel. Les phases organiques réunies sont lavées avec une solution saturée d'hydrogénocarbonate de sodium (120 mL), puis avec de la saumure (100 mL), séchées sur sulfate de magnésium, filtrées et concentrées sous vide partiel.
Les phases organiques réunies sont lavées avec une solution saturée de bicarbonate de sodium (60 mL), puis de la saumure (50 mL), séchées sur sulfate de magnésium, filtrées et concentrées sous vide partiel.

FONCTIONNALISATION DE DIOLS BICYCLIQUES
- Métathèse pallado-catalysée du cycloocta-1,5-diène : accès aux précurseurs bicycliques
- Mécanisme de diacétoxylation transannulaire pallado-catalysée du cycloocta-1,5-diène
- Hydrolyse alcaline du diacétoxy[3.3.0]bicyclooctane
- Résolution enzymatique du (1R*,3aS*,4R*,6aS*)-octahydropentalène-1,4-diol racémique
- Résolution enzymatique
- Détermination des excès énantiomériques
- Stratégie silyle : O-monosilylation des (1R*,3aS*,4R*,6aS*)-octahydro-pentalène-1,4-diols
- Stratégie benzyle : O-monobenzylation des (1R*,3aS*,4R*,6aS*)-octahydro-pentalène-1,4-diols
- Conclusion
Cette métathèse fournit ensuite du cis-endo-endo-bicyclo[3.3.0]octane 23 racémique diacétoxylé avec un rendement de 75 %. L'hydrolyse alcaline du diester racémique 23 obtenu précédemment fournit le C2-diol symétrique, le diendo-cis-bicyclo[3.3.0]octane-2,6-diol racémique 24. Nous avons maintenant deux diendo-cis-bicyclo[3.3.0 ]octane-2, 6-Enantiopure diols 24A et 24B et racémique diol 24, qui doivent être fonctionnalisés sous forme d'hydroxy-pentalène-octahydro-1-ols monoprotégés.
Schéma 113 : Stratégie envisagée pour la synthèse de cis-jasmonates de méthyle à partir de diendo-cis-bicyclo[3.3.0]octane-2,6-diols monoprotégés.
![Tableau 15 : Résultats obtenus pour la résolution enzymatique du diendo-cis-bicyclo[3.3.0]octane-2,6-diol racémique](https://thumb-eu.123doks.com/thumbv2/1bibliocom/465302.70576/188.892.107.777.295.430/tableau-résultats-obtenus-résolution-enzymatique-diendo-bicyclo-racémique.webp)
Oxydation des (1S*,3aR*,4S*,6aR*)-octahydro-pentalène-1,4-diols O-silylés et O-benzylés
Résolution enzymatique de la (3aR*,6aR*)-octahydro-pentalène-1,4-dione en présence d’une alcool
Débenzylation des (3aR*,4S*,6aR*)-4-benzyloxy-octahydro-pentalène-1-ones par hydrogénolyse
Silylation des (3aR*,4S*,6aR*)-4-hydroxy-octahydro-pentalène-1-ones
Homologation régiosélective de silanyloxycétones bicycliques
- Homologation simple de cétones et d’aldéhydes
- Homologation régiosélective des (3aR*,4S*,6aR*)-4-silanyloxy-octahydro-pentalène-1-ones
- Mécanisme d’homologation régiosélective des (3aR*,4S*,6aR*)-4-silanyloxy-octahydro-pentalène-1-
ÉNOLISATION ET COUPURE OXYDANTE DE
Enolisation régiosélective des (3aR*,4S*,6aR*)-4-silanyloxy-octahydro-pentalèn-1-ones silylées et
Enolisation régiosélective d’une (1S*,3aS*,7aR*)-silanyloxy-octahydro-indèn-5-one racémique
Coupure oxydante d’éthers d’énols silylés
- Coupure oxydante d’énolates de silyle non-homologués
- Coupure ozonolytique d’énolates de silyle homologués
Préparation de jasmonoïdes clé
- Réaction de Wittig, déprotection, et lactonisation intramoléculaire : préparation de la (±)- δ -jasmolactone
- Préparation d’halogénures d’alkylphosphonium par irradiation sous champ micro-onde
- Synthèse de la (±)- δ -jasmolactone
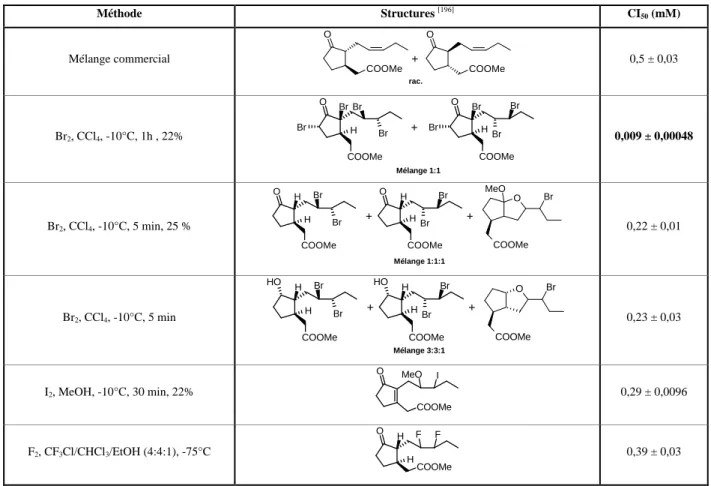
- Synthèse du cis-jasmonate de méthyle racémique et degré d’épimérisation
Conclusion


![Figure 7 : Voies de signalisations intrinsèques apoptotiques au sein des canaux ioniques mitochondriaux en phase pré-apoptotique (gauche) et post-apoptotique (droite) [156]](https://thumb-eu.123doks.com/thumbv2/1bibliocom/465302.70576/56.892.101.779.391.729/figure-signalisations-intrinsèques-apoptotiques-ioniques-mitochondriaux-apoptotique-apoptotique.webp)