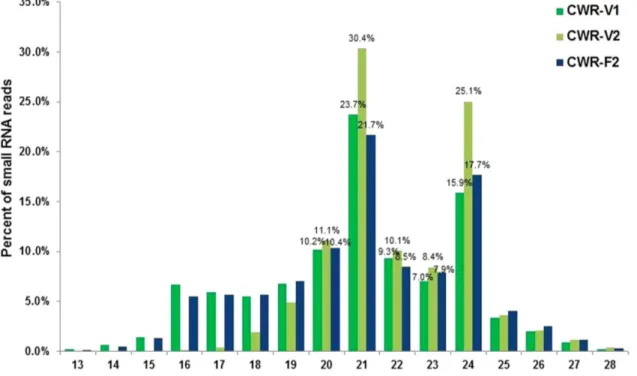
Identification and functional analysis of flowering related microRNAs in common wild rice (Oryza rufipogon Griff.).
Texto
Imagem

Documentos relacionados
canephora (Combes et al. Since there are no coffee miRNA se- quences in the current release of the miRBase database, all miRNAs identified here are new for the refereed species.
This was observed in a study in Sri Lanka rice fields in which the richness and abundance of species and the diversity of all sampled arthropod groups, except Diptera,
14 Uwe Johnson, 1934 geboren, gehört zu einer Generation, die NS-Zeit und Krieg nur als Kind und Heranwachsender erlebt hat, jünger noch als Günter Grass, der hier
Para este estágio parcelar, além dos objetivos propostos pela UC, delineei alguns objetivos pessoais, como conduzir consultas autonomamente, ganhando confiança para as
Arabidopsis and rice, and the mature RC-miRNAs with normalized total read counts more than 20 RPM (reads per million) in other plant species (the mature RC- miRNAs of soybean
After aligning the sequencing reads from Ae.aegypti saliva to the Ae.aegypti miRNA database, 72 distinct known miRNAs were identified in both uninfected and CHIKV-infected
CONCEPÇÕES E PRÁTICAS O DILEMA DA AVALIAÇÃO DA APRENDIZAGEM: Um estudo de caso da prática avaliativa de professores da rede estadual de ensino do Maranhão Dissertação apresentada
Relative expression estimates for the target miRNAs (miRNA-21, miRNA-125b, miRNA-145, and miRNA-630) were obtained by image analysis of the ISH-stained slides ( Table 1 ).The
