Journal of the Renin- Angiotensin-Aldosterone System
(Including other peptidergic systems)
March 2001 Volume 2
Role of basic amino acids of the human angiotensin
type 1 receptor in the binding of the non-peptide
antagonist candesartan
Georges Vauquelin,* Frederik LP Fierens,* Zsuzsanna Gáborik,† Tam Le Minh,*
Jean-Paul De Backer,* László Hunyady,† Patrick ML Vanderheyden*
Keywords: CHO cells, angiotensin II, human AT1-receptor, mutation, candesartan, insurmountable, non-peptide antagonist
*Department of Molecular and Biochemical Pharmacology, Institute for Molecular Biology and Biotechnology, Free University of Brussels (VUB), B-1640 Sint-Genesius Rode,
Belgium
†Department of
Physiology, Semmelweis University Medical School, H-1444 Budapest, PO Box 259, Hungary
Correspondence to: Professor G Vauquelin Department of Molecular and Biochemical Pharmacology, Institute for Molecular Biology and Biotechnology, Free University of Brussels (VUB), Paardenstraat 65, B-1640 Sint-Genesius Rode,
Belgium
Tel: +32 2 358 3139 Fax: +32 2 359 0276 E-mail: gvauquel@ vub.ac.be
JRAAS2001;2 (suppl 1):S32-S36
Abstract
To explain the insurmountable/long-lasting binding of biphenyltetrazole-containing AT1-receptor antagonists
such as candesartan, to the human angiotensin II type 1-receptor, a model is proposed in which the basic amino acids Lys199and Arg167of the receptor interact
respectively with the carboxylate and the tetrazole group of the antagonists. To validate this model, we have investigated the impact of substitution of Lys199by Ala or
Gln and of Arg167by Ala on the binding properties of
[3H]candesartan and on competition binding by
candesartan, EXP3174, irbesartan, losartan, angiotensin II (Ang II) and [Sar1-Ile8]angiotensin. Our results
indicate that both amino acids play an important role in the AT1-receptor ligand binding. Whereas the negative
charge of Lys199is involved in an ionic bond with the
end-standing carboxylate group of the peptide ligands, its polarity also contributes to the non-peptide antagonist binding. Substitution of Arg167by Ala
completely abolished [3H]Ang II, as well as [3H]
candesartan, binding. Whereas these results are in line with the proposed model, it cannot be excluded that both amino acid residues are important for the structural integrity of the AT1-receptor with respect to its ligand
binding properties.
Introduction
The therapeutic use of selective, non-peptide antagonists for the angiotensin II type 1 (AT1 )-receptor in the treatment of hypertension is well established.1 Experiments with Chinese Hamster Ovary cells, which have been permanently trans-fected with the gene coding for the human AT1
-receptor (CHO-hAT1cells) have shed new light on
the molecular mechanism of action of these AT1
-receptor antagonists. One of the major findings is that these antagonists exhibit a competitive type of interaction with angiotensin II (Ang II) on the receptor: they bind to a common or overlapping site on the receptor in a mutually exclusive way.2 The ability of some of these antagonists to decrease the maximal effect of Ang II in vascular smooth muscle contraction studies, a phenome-non denominated as ‘insurmountable’ inhibition, is merely the result of their slow dissociation from the receptor and the experimental set-up.3,4Since these functional studies involve a preincubation of the tissue with the antagonist, the blockade of the receptors may be sufficiently long-lasting to
prevent their access to subsequently-added Ang II. Hence, maximal receptor stimulation may not be achieved during the relatively short challenge of the tissue with the agonist. In contrast,‘surmount-able’ antagonists, such as losartan, undergo rapid dissociation from the AT1-receptor and hence only produce parallel rightward shifts of the Ang II dose-response curve.5,6
The extent by which ‘insurmountable’ antago-nists depress the maximal response to Ang II is highly variable; it is almost complete for candesar-tan5,7but only partial for irbesartan, valsartan and EXP3174.3,8-10 To explain these findings, it was proposed that antagonist/AT1-receptor complexes might adopt two distinct states: a fast reversible state, which accounts for the surmountable inhi-bition, and a tight-binding state, which accounts for the insurmountable inhibition.4The simplest means to explain these binding properties is a two-state, two-step model, in which the initial binding of all antagonists is fast and reversible.To achieve insurmountable inhibition, the antagonist-receptor complex must further be converted into a tight binding state. Whereas the extent of this conversion is variable for the different inmountable antagonists, it cannot occur for sur-mountable antagonists, such as losartan.
The structure of insurmountable antagonists such as candesartan, EXP3174, valsartan and irbe-sartan is based on the biphenyltetrazole substruc-ture of losartan.11 It is therefore likely that their ability to form tight-binding complexes with the AT1-receptor is dictated by the chemical nature of
Journal of the Renin- Angiotensin-Aldosterone System
(Including other peptidergic systems)
March 2001
report, it was found that the substitution of Lys199 into Gln produces a more pronounced decrease of the binding affinity of insurmountable antagonists when compared with losartan.19Here, we set out to investigate the effects of more rigorous substi-tutions of Lys199and Arg167into Ala on the binding of losartan and insurmountable antagonists. A model for antagonist/AT1-receptor interaction,
combining the present data with the previously-published working hypotheses is presented.
Materials and methods
Materials
Candesartan,7EXP3174,10losartan10and irbesartan8 were obtained from AstraZeneca (Mölndal, Sweden). Ang II and [Sar1-IIe8]angiotensin were obtained from Sigma. [3H]candesartan (17 Ci/mmol) was kindly provided by AstraZeneca.
Lipofectamine was from GibcoBRL, Life
Technologies, Merelbeke, Belgium. All other chemicals were of the highest grade commercially available.
Mutagenesis of human AT1-receptor DNA,
cell culture, transient transfection and
[3H]candesartan binding
The cloning of the human AT1-receptor cDNA and the site-directed mutagenesis has been described previously.19 Wild Type Chinese Hamster Ovary Cells (CHO-K1), that were kindly provided by Dr H Verschueren (Pasteur Institute, Brussels, Belgium), are transiently transfected in 12-well plates using Lipofectamine (Life Technologies) as described before. Two days after transfection, the [3H]candesartan binding assay was carried out on adherent intact cells. For this purpose, cells were
washed twice with DMEM (1 ml/well), and then incubated for 60 minutes at 37°C with [3 H]can-desartan at 5 nM for competition binding and kinetic experiments, or with a concentration range between 0.5 and 50 nM for saturation binding. Non-specific binding was measured in the presence of 1 µM unlabelled candesartan and was subtracted from total binding to yield specific binding. The cell-bound radioactivity was extracted and measured as described before.6
Calculations
The binding parameters from the dissociation, saturation and competition binding experiments (k-1, KD and IC50 values) were calculated by non-linear regression analysis using GraphPad Prism (San Diego, CA, USA), based on a one-site bimolec-ular reaction obeying the mass law action.A com-petitive type of interaction between [3H] candesartan and the other antagonists and Ang II was described previously.2At the highest concen-trations used, the unlabelled antagonists produced full inhibition of the specific [3H]candesartan binding in the competition experiments. The Ki values were calculated from the IC50values by the Cheng and Prusoff equation.20
Results
CHO-K1 cells are transiently transfected with the gene coding for the wild-type human AT1-receptor
(CHO-WT AT1).The impact of replacing Lys199 with Gln (CHO-Lys199Gln) or with Ala (CHO-Lys199Ala), or replacing Arg167 with Ala (CHO-Lys199Ala), on the binding properties of selective non-peptide antag-onists in [3H]candesartan binding assays on intact adherent cells was studied.The specific binding of
Figure 1 Competition binding curves of AT1-receptor
ligands on CHO-Lys199Ala cells incubated for 60 minutes at
37°C with 5 nM [3H]candesartan and increasing
concentrations of candesartan (), EXP3174 (), irbesartan (), losartan (), [Sar1-Ile6]Ang (
r) or angiotensin II (▲). Data points are the average±SEM of three independent experiments.The corresponding equilibrium dissociation constants are given as pKi values in Table 1. Insert:A typical saturation curve of [3H]
candesartan binding to CHO-Lys199Ala cells. Specific
binding (cpm/well) is expressed as function of the free radioligand concentration (nM).
Figure 2 Dissociation rate of [3H]candesartan binding.
The radioligand was incubated at a concentration of 1.5 nM with CHO-WT AT1() or with CHO-Lys199Gln cells
(▲) and of 5 nM with CHO-Lys199Ala (
r) cells until equilibrium. Then 1 µM unlabelled candesartan was added to initiate dissociation.The dissociation rate constants (k-1
values) were determined by non-linear regression analysis and are 0.0037±0.00018, 0.019±0.00094 and 0.215±0.014 minutes-1for the WT, Lys199Gln and the Lys199Ala receptor
respectively.The data points are the average±SEM of three independent experiments.
Antagonist concentration (log M) Dissociation time (min)
[
3H]-candesartan binding
(% of contr
ol)
[
3H]-candesartan binding
(% of contr
Journal of the Renin- Angiotensin-Aldosterone System
(Including other peptidergic systems)
March 2001 Volume 2 Supplement 1
[3H]candesartan was time-dependent and reached equilibrium after 30 minutes at 37°C at a radioli-gand concentration of 1.5 nM for CHO-WT AT1
and CHO-Lys199Gln cells and at 5 nM for CHO-Lys199Ala cells. The calculated first order associa-tion rate constants (k1values) were 0.175±0.118, 0.0286±0.0087 and 0.0259±0.0078 nM-1 minute-1 respectively. No specific binding of [3 H]candesar-tan or [3H]Ang II was observed for the CHO-Arg167Ala cells (data not shown). The equilibrium dissociation constant (KD) on the CHO-Lys199Ala cells was determined by non-linear regression analysis of saturation binding curves and was 45±9 nM (Figure 1).
The dissociation rate of [3H]candesartan from CHO-WT AT1, CHO-Lys199Gln and CHO-Lys199Ala cells was calculated from the kinetic experiments shown in Figure 2. As shown in this figure, the dis-sociation rate of [3H]candesartan was increased in CHO-Lys199Gln cells and even further in CHO-Lys199Ala cells.
Competition binding curves with 5 nM [3H]candesartan to CHO-Lys199Ala cells (Figure 1) yielded IC50values from which the corresponding Ki values of the non-peptide AT1 antagonists and
Ang II were determined; i.e., 32.0, 63.3, 40.4, 521, 8334 and 416 nM respectively for candesartan, EXP3174, irbesartan, losartan, Ang II and [Sar1 -Ile8]Ang. They are compared with the values for the wild-type receptor, as well as previously reported Ki values on CHO-Lys199Gln cells19 (Table 1).
Discussion
Pharmacological studies with candesartan and its analogues revealed that these antagonists only produce insurmountable inhibition of the AT1
-receptor when they possess a carboxyl group at well-defined positions.7In addition, the design of EXP3174, another losartan-like biphenyltetrazole-containing AT1-receptor antagonist, has been
based on the premise that its carboxyl group should point to the same positive charge of the receptor as the carboxyl-terminus of Ang II.11,21In this respect, earlier mutation studies revealed that the carboxyl-terminus of Ang II interacts with the side chain of Lys199of the AT
1-receptor.19,22,23Based
on these considerations, a model was proposed, in which the tight/insurmountable binding of antag-onists such as candesartan and EXP3174 might
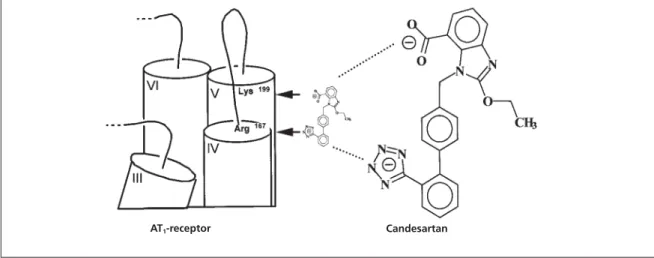
result from strong electrostatic interactions between their negatively-charged tetrazole and carboxyl groups with basic amino acid residues of the AT1-receptor. Further support for this hypothe-sis comes from the fast reversible/surmountable binding of losartan, which contains only a single negatively-charged tetrazole moiety. Taken together, there are good grounds to anticipate that Lys199 plays a pivotal role for the slow reversible binding of non-peptide antagonists such as EXP3174 and candesartan. Consequently, the tetrazole moiety of antagonists should interact with another basic amino acid of the receptor and, in this respect, the side-chain of Arg167 has been proposed as a likely candidate.24A graphical representation that integrates these considera-tions is given in Figure 3.
To investigate the possible role of basic amino acids of the AT1-receptor for agonist and
antago-nist binding, numerous mutagenesis studies have been reported. However, the conclusions of these studies are limited as they are mainly focused on the effect of the receptor structure on the binding properties of Ang II, the surmountable antagonist losartan or on peptide antagonists. Only limited attention has been paid to compare the effect of such substitutions on the binding properties of insurmountable antagonists.25,26 The availability of [3H]candesartan, an antagonist capable of under-going tight, long-lasting binding to AT1-receptors, now permits the direct evaluation of the impact of such receptor mutations on insurmountable antagonist-receptor interactions. Moreover, because of its high affinity and extremely low non-specific binding, this radioligand allows the study of AT1-receptors in intact cells instead of the hitherto widely-used cell membranes.6,25,26 The present work has been carried out on wild-type and mutated human AT1-receptors and has been
undertaken to verify the potential implication of Lys199 and Arg167 in the fast and slowly reversible (i.e., surmountable and insurmountable) binding of the non-peptide AT1-receptor antagonists,
can-desartan, EXP3174, irbesartan and losartan. The impact of replacing Lys199by Gln or Ala on the binding properties of the AT1-receptor was determined. Although the side-chain of Gln is uncharged, its amide function is still polar and is potentially able to form inter- and/or intramolecu-lar hydrogen bonds. As compared with the
wild-Table 1 pKi values of AT1-receptor ligands for CHO-Lys199Ala cells calculated from competition curves with [3H]
candesartan (Figure 2). Data are compared with values for CHO-WT AT1or CHO-Lys199Gln cells reported previously.19
Ligand pKi Lys199Ala Ki(Lys199Ala)/Ki(WT) Ki(Lys199Ala)/Ki(Lys199Gln)
Angiotensin II 5.03±0.11 1204 1.1
[Sar1-Ile8]Angiotensin 6.38±0.08 2625 2.9
Candesartan 7.49±0.07 1189 26
EXP3174 7.20±0.07 1417 80
Irbesartan 7.39±0.09 386 40
Journal of the Renin- Angiotensin-Aldosterone System
(Including other peptidergic systems)
March 2001
type receptor, the Lys199Gln mutant displayed a more than 1000-fold reduction of the affinity of Ang II and the structurally-related antagonist [Sar1,Ile8]angiotensin.24No further decrease of the affinity of both peptides was observed for the Lys199Ala mutant (Table 1).These data indicate the unequivocally important contribution of a posi-tively-charged Lys199residue in the binding of these peptides. When replaced by a non-charged amino acid, it is no longer able to contribute in the binding energy of these ligands.
In contrast, the binding affinity for the non-peptide antagonists was less severely reduced by the Lys199 to Gln mutation, yet it was more pro-nounced for the insurmountable antagonists, candesartan and EXP3174, as compared with losartan.19 The Lys199 to Ala mutation caused a dramatic increase of the dissociation rate of [3H]candesartan and a substantial decrease of the affinity for all the investigated non-peptide antag-onists.The 400-fold reduction in losartan affinity is not compatible with the 12- to 14-fold reduction reported by Noda et al. for the Lys199 to Ala mutation.27 This might be ascribed to species-related differences in the receptor structure (rat AT1A vs. human AT1-receptor), or by the use of
membrane preparations instead of intact cells that might affect the antagonist-receptor interaction. When the findings of both mutations are taken together, it appears that both the polarity of Lys199 and its ability to form a hydrogen bond play a sub-stantial role in the formation of the non-peptide-antagonist/receptor complex. However, the relative contribution of an ionic bound is less important than for peptide ligands.Taken together, the presence of Lys at position 199 may, in addition to its importance for the tight binding of antagonists to the receptor, also play a structural role in keeping the receptor in an optimal confor-mation for the binding of non-peptide antagonists. The present findings suggest that only this latter property is exerted by Gln at this position.
Specific binding of [3H]candesartan and [3H]Ang II was completely lost when Arg167of the human AT1-receptor was replaced by Ala.This is in
agreement with earlier observations24,27 and com-patible with the proposal by Yamano et al.24that the anionic tetrazole moiety of the non-peptide AT1-receptor antagonists interacts with Arg167 in
the wild-type receptor. In the same way, the loss of [3H]Ang II binding in the Ala167 mutant could be explained in terms of a potential interaction between Arg167 and Tyr4of the agonist.24An alter-native interpretation of the Arg167mutation is that this amino acid may play a structural and/or con-formational role with respect to agonist and antag-onist binding. In this context, it is noteworthy that Lys199 and Arg167 are located at the interface between transmembrane segments IV and V and their joining extracellular loop (Figure 3). Because of the crucial location of these polar amino acids, it cannot be excluded that their mutation into nonpolar ones, such as Ala, could affect the sepa-ration and/or tilting between segments IV and V and thereby provoke a structural disorganisation of the receptor.
In conclusion, our mutation studies are com-patible with a rudimentary working hypothesis (Figure 3) in which the tetrazole moiety of non-peptide AT1-receptor antagonists interacts with
the side-chain of Arg167of the receptor, while Lys199 may be involved in the formation of a tight binding/slow reversible state of the antagonist-receptor complex. However, it cannot yet be excluded that these amino acid residues are also involved in the structural integrity of the AT1
-receptor with respect to its ligand binding properties.
Acknowledgements
We are most obliged to AstraZeneca Sweden,Astra Belgium and the Queen Elisabeth Foundation, Belgium, for their kind support. We thank Drs S Meloche and G Guillemette for kindly supplying us the cDNA of the human AT1-receptor.This text
presents research results of the Belgian program on Interuniversity Poles of Attraction initiated by the Belgian State, Prime Minister’s Office, Science Policy Programming. The scientific responsibility is assumed by its authors.
Figure 3 Graphical representation of the possible interaction of Arg167with the tetrazole moiety and of Lys199with the
carboxylate group of candesartan.
Journal of the Renin- Angiotensin-Aldosterone System
(Including other peptidergic systems)
March 2001 Volume 2 Supplement 1
References
1. Vallotton MB. The renin-angiotensin system. Trends
Pharmacol Sci1987;8:69-74.
2. Vanderheyden PML, Fierens FLP, De Backer JP,Vauquelin G. Reversible and syntopic interaction between angiotensin II AT1
receptor antagonists, human AT1receptors expressed in
CHO-K1 cells.Biochem Pharmacol2000;59:927-35.
3. Criscione L, de Gasparo M, Bühlmayer P, Whitebread S, Ramjoué HP, Wood J. Pharmacological profile of valsartan: a potent, orally active, non-peptide antagonist of the angiotensin II AT1receptor subtype.Br J Pharmacol. 1993;110:761-71.
4. Fierens FLP, Vanderheyden PML, De Backer J-P Vauquelin G. Insurmountable angiotensin II AT1receptor antagonists: the
role of tight antagonist binding. Eur J Pharmacol
1999;372:199-206.
5. Vanderheyden P, Fierens FLP, De Backer J-P, Frayman N, Vauquelin G. Distinction between surmountable and insur-mountable selective AT1receptor antagonists by use of
CHO-K1 cells expressing human angiotensin II AT1receptors.Brit J
Pharmacol1999;126:1057-65.
6. Fierens FLP, Vanderheyden PML, De Backer J-P Vauquelin G. Binding of the antagonist [3H]candesartan to angiotensin II
AT1 receptor-transfected Chinese hamster ovary cells.Eur J
Pharmacol 1999;367:413-22.
7. Noda M, Shibouta Y, Inada Y et al.Inhibition of rabbit aoric angiotensin II (AII) receptor by CV-11974, a new nonpeptide AII antagonist.Biochem Pharmacol1993;46:311-8.
8. Cazaubon C, Gougat J, Bousquet F et al.Pharmacological characterization of SR 47436, a new non-peptide AT1subtype
angiotensin II receptor antagonist. J Pharmacol Exp Ther
1993;265:826-34.
9. Verheijen I, Fierens FLP, De Backer JP, Vauquelin G, Vanderheyden PML. Interaction between the partially insur-mountable antagonist valsartan, human recombinant angiotensin II type 1 receptors. Fund Clin Pharmacol
2001;14:577-85.
10. Wong PC, Price WA, Chiu AT, Thoolen MJMC et al. Non-peptide angiotensin II receptor antagonists. IX. Pharmacology of EXP3174: an active metabolite of DuP 753, an orally active antihypertensive agent.J Pharmacol Exp Ther
1990;255:211-7.
11. Timmermans PBMWM, Wong PC, Chiu AT et al. Angiotensin II receptors, angiotensin II receptor antagonists.
Pharmacological Rev1993;45:205-51.
12. Robertson MJ, Barnes JC, Drew GM et al. Pharmacological profile of GR 117289 in vitro: a novel, potent and specific non-peptide angiotensin AT1-receptor antagonist.Br J Pharmacol
1992;107:1173-80.
13. Tamura K, Okuhira M, Mikoshiba I, Hashimoto K.In vitro
pharmacological properties of KRH-594, a novel angiotensin II type 1 receptor antagonist.Biol Pharmacol Bull1997;20:850-5.
14. Dickinson KE, Cohen RB, Skwish S et al. BMS-180560, an insurmountable inhibitor of angiotensin II-stimulated responses: comparison with losartan and EXP3174. Br J
Pharmacol1994;113:179-89.
15. Panek RL, Lu GH, Overhisser RW, Major TC, Hodges JC, Taylor DG. Functional studies but not receptor binding can dis-tinguish surmountable from insurmountable AT1 antagonism.
J Pharm Exp Ther1995;273:753-61.
16. De Arriba AF, Gomez-Casajus LA, Cavalcanti F, Almansa C, Garcia-Rafanel J, Forn J.In vitropharmacological characteriza-tion of a new selective angiotensin AT1receptor antagonist,
UR-7280.Eur J Pharmacol1996;318:341-7.
17. Yu SS, Lefkowitz RJ, Hausdorff WP.β-Adrenergic receptor sequestration: a potential mechanism of receptor resensitisa-tion.J Biol Chem1993;268:337-41.
18. Renzetti A, Cucci P, Guelfi M, Cirillo R, Salimbeni A, Giachetti A. Pharmacology of LR-B/057, a novel orally active AT1
receptor antagonist.J Cardiovasc Pharmacol1995;25: 354-60. 19. Fierens FLP, Vanderheyden PML, Gaborik Z et al. Lys199 Mutation of the human angiotensin II AT1receptor
differential-ly affects the binding of surmountable, insurmountable non-peptide antagonists.JRAAS 2000;1:283-8.
20. Cheng YC, Prusoff WH. Relationship between the inhibi-tion constant (Ki) and the concentrainhibi-tion of inhibitor wich causes 50% inhibition (IC50) of an enzymatic reaction.Mol
Pharmacol1973;32:2497-503.
21. Duncia JV, Chiu AT, Carini DJ et al.The discovery of potent non-peptide angiotensin II receptors antagonists: a new class of potent antihypertensives.J Med Chem1990;33:1312-29. 22. Inoue Y, Nakamura N, Inagami T. A review of mutagenesis studies on angiotensin II type 1 receptor, the three-dimension-al receptor model in search of the agonist and antagonist binding site and the hypothesis of a receptor activation mechanism, J Hypertens1997;15:703-14.
23. Mirua SI, Feng Y-H, Huasain A, Karnik SS. Role of aromatic-ity of agonist switches of angiotensin II in the activation of the AT1receptor.J Biol Chem1999;274:7103-10.
24. Yamano Y, Ohyama K, Kikyo M et al. Mutagenesis and the molecular modeling of the rat angiotensin II receptor (AT1).
J Biol Chem1995;270:14024-30.
25. Schambye HT, Hjorth SA, Bergsma DJ, Sathe G, Schwartz TW. Differentiation between binding sites for angiotensin II and non-peptide antagonists on the angiotensin II type 1 receptors.Proc Natl Acad Sci USA1994;91:7046-50. 26. Schambye HT, van Wijk B, Hjorth SA et al.Mutations in transmembrane segment VII of the AT1receptor differentiate
between closely related insurmountable and competitive angiotensin antagonists.Br J Pharmacol1994;113:331-3. 27. Noda K, Saad Y, Karnik SS. Interaction of Phe8 of angiotensin II with Lys199 and His256 of AT1 receptor in
![Figure 1 Competition binding curves of AT 1 -receptor ligands on CHO-Lys 199 Ala cells incubated for 60 minutes at 37°C with 5 nM [ 3 H]candesartan and increasing concentrations of candesartan (), EXP3174 (), irbesartan (), losartan (), [Sar 1 -Ile 6 ]An](https://thumb-eu.123doks.com/thumbv2/123dok_br/18153591.327875/2.892.179.823.264.556/competition-incubated-candesartan-increasing-concentrations-candesartan-irbesartan-losartan.webp)
![Table 1 pKi values of AT 1 -receptor ligands for CHO-Lys 199 Ala cells calculated from competition curves with [ 3 H]](https://thumb-eu.123doks.com/thumbv2/123dok_br/18153591.327875/3.892.175.830.996.1168/table-values-receptor-ligands-cells-calculated-competition-curves.webp)