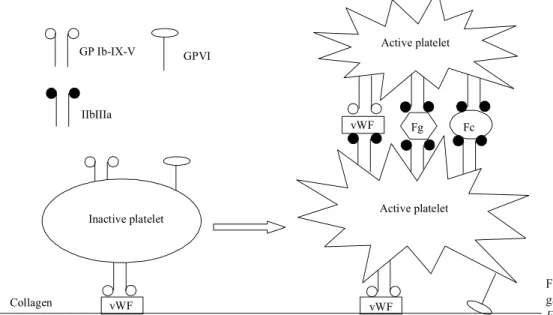
Novel aspects of platelet aggregation
Texto
Imagem
Documentos relacionados
A pro- jecção dos valores da média e do desvio padrão, no gráfico de correlação defi- nido pelo mesmo autor (1969), indica para 40% das amostras valores típicos das areias de
The main objective of this work plan is to develop an efficient downstream processing procedure for Tetraselmis sp. CTP4, comprising an efficient extraction
The pathophysiology of thrombus formation in the portal system is still unclear. Admittedly, there may be a combination between prethrombotic factors and local factors. Local
The increase in relative vWF activity in COPD patients, when compared with the smoker group, suggests that vWF may play a role in the inflammatory pathophysiology of COPD and
8 In fact, AT may be able to exert an antiarrhythmic effect and other effects on RR interval because the higher frequency power (HF) of HRV, vagal modulation, suggests that
In fact, the participation of NER proteins in the removal of oxidized DNA bases suggests a crosstalk between NER and other DNA repair pathways, and Pol eta may also protect cells
mice infectivity and biofilm formation in the tested conditions, prophages.. encoding platelet-like binding proteins promote adhesion
Several extracellular matrix proteins including fibronectin, throm- bospondin-1, laminin, SPARC, and osteopontin have been suggested to modulate tumor phenotype by affecting
