Hana Zouk1,2, Luc Marchand1, Constantin Polychronakos1,2,3*
1Endocrine Genetics Laboratory, McGill University Health Center, Montreal Children’s Hospital Research Institute, McGill University, Montreal, Quebec, Canada, 2Department of Human Genetics, McGill University, Montreal, Quebec, Canada,3Department of Paediatrics, McGill University Health Centre, Montreal, Quebec, Canada
Abstract
Background:The Thr allele at the non-synonymous single-nucleotide polymorphism (nsSNP) Thr946Ala in theIFIH1gene confers risk for Type 1 diabetes (T1D). The SNP is embedded in a 236 kb linkage disequilibrium (LD) block that includes four genes:IFIH1,GCA,FAPandKCNH7. The absence of common nsSNPs in the other genes makes theIFIH1SNP the strongest functional candidate, but it could be merely a marker of association, due to LD with a variant regulating expression levels of IFIH1or neighboring genes.
Methodology/Principal Findings:We investigated the effect of the T1D-associated variation on mRNA transcript expression of these genes. Heterozygous mRNA from lymphoblastoid cell lines (LCLs), pancreas and thymus was examined by allelic expression imbalance, to detect effects incison mRNA expression. Using single-nucleotide primer extension, we found no difference between mRNA transcripts in 9 LCLs, 6 pancreas and 13 thymus samples, suggesting thatGCAandFAPare not involved. On the other hand, KCNH7 was not expressed at a detectable level in all tissues examined. Moreover, the association of the Thr946Ala SNP with T1D is not due to modulation ofIFIH1expression in organs involved in the disease, pointing to theIFIH1nsSNP as the causal variant.
Conclusions/Significance:The mechanism of the association of the nsSNP with T1D remains to be determined, but does not involve mRNA modulation. It becomes necessary to study differential function of theIFIH1protein alleles at Thr946Ala to confirm that it is responsible for the disease association.
Citation:Zouk H, Marchand L, Polychronakos C (2010) Study of Transcriptional Effects inCis at theIFIH1 Locus. PLoS ONE 5(7): e11564. doi:10.1371/ journal.pone.0011564
Editor:Adrian Vella, Mayo Clinic College of Medicine, United States of America
ReceivedApril 25, 2010;AcceptedJune 14, 2010;PublishedJuly 13, 2010
Copyright:ß2010 Zouk et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Funding:This work was funded by the Juvenile Diabetes Research Foundation International. HZ is supported by a doctoral scholarship from the Fonds de Recherche en Sante du Quebec. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests:The authors have declared that no competing interests exist.
* E-mail: [email protected]
Introduction
Type 1 diabetes (T1D) is a complex disease involving both genetic and environmental factors. This is largely attributed to genetic variation among individuals at several loci. One of them involves theIFIH1gene (interferon-induced helicase 1) where the Thr allele at the Thr946Ala polymorphism increases T1D risk [1]. The associated SNP (rs1990760) is embedded in a 236 kb linkage disequilibrium (LD) block on Chr 2q24.3 that includes four genes:
IFIH1, also known ashelicardorMDA-5(melanoma differentiation associated gene-5); GCA (grancalcin); FAP (fibroblast activation proteinasubunit) andKCNH7(potassium voltage gated channel subfamily H7), any of which could harbor a T1D-associated functional variant. IFIH1 belongs to a family of RNA helicases that bind double-stranded viral RNA [2,3] and induces a type I interferon anti-viral response [4]. This is particularly interesting given the evidence for a role of enteroviruses in the etiology of T1D [5–7]. The recent discovery of rare nsSNPs in IFIH1
protective of T1D [8], two of which involve loss of function [9], strongly supports it as the gene involved, but locus heterogeneity remains a possibility given that thousands of loci with weak effects likely account for each complex trait [10].GCAencodes a calcium binding protein that is expressed in most immune cells and is associated with degranulation, and consequently, immune reaction [11]. FAPencodes a human stromal antigen, which can in turn
activate a T-cell mediated cellular response [12].KCNH7encodes a potassium voltage-gated channel which has many roles, most notably, the regulation of insulin secretion [13]. Hence, all genes at the IFIH1 locus may be interesting potential candidates in the etiology of T1D even though nsSNPs are more likely to have functional effects. Therefore, theIFIH1nsSNP obviously remains the strongest functional candidate; however, the fact that it could be merely a marker of association that tags another variant regulating expression levels ofIFIH1or of neighboring genes, must be ruled out. In the same paper that reported the functional effect of the rareIFIH1SNPs, the Thr946Ala nsSNP was not found to have any effect of protein function [9]. However, because a transfection system was used, where IFIH1 alleles are over-expressed, small differences between them may not be detectable. Allele-dependent specificities for sequence, length, or other characteristics of specific viral RNAs would also not have been detected.
400,000 SNPs, no transcriptional effects in ciswere observed on theIFIH1locus or any of the surrounding regions [15]. In another paper using the same methodology as Liuet al.[14],IFIH1allelic expression differences were not observed [16]. This inconsistence in reported results could be due to the fact that both groups compared mRNA levels among individuals of different genotypes. This has the disadvantage of introducing a large amount of background noise from different individuals’ immune experience, loci in trans [17], quality of RNA extraction (including mRNA degradation), assay variance, loading normalization, etc., making it difficult to properly detect the typically small functional effects seen at complex-trait loci. A much more robust approach to measure mRNA variation, known as allelic expression imbalance (AI), removes this noise by comparing expression levels of the two alleles originating from the same individual in samples heterozy-gous for a transcribed SNP. This method has been well validated in our laboratory [18,19], and others [17,20–23].
AI could stem from allele-dependent effects of one or more polymorphisms incisthat alter promoter function as well as that of other regulatory elements such as enhancers and silencers, potentially affecting transcription or RNA stability. Thus, the presence of a regulatory polymorphism that is in LD with the marker SNP could result in AI [21].
In AI assays, each allele acts as an internal control for confounding factors that alter the overall expression of the gene in question, thus maximizing the sensitivity of detecting effects in
cison mRNA expression in the same RNA sample, from the same individual. In samples that are heterozygous for a cis-acting regulatory variant, one allele will be expressed at a higher level than the other [21,24,25]. Heterozygous genomic DNA from the same source is used as a control for 1:1 stoichiometry [24]. It is worth noting that another advantage of AI is that this assay relies on the comparison of allelic ratio in DNA and mRNA of each individual. Thus, it automatically controls for any polymorphisms present in the primer sites or copy number variation encompassing the gene studied, which normally exert a similar effect on both DNA and mRNA [26].
The purpose of this study was to investigate the effect of the T1D-associated variation on mRNA expression ofIFIH1 and all other genes in the LD block by allelic expression imbalance, using single-nucleotide primer extension (SNuPE) on RT-PCR products of heterozygous lymphocyte, thymic and pancreatic RNA samples, to cover tissues most important in T1D.
Materials and Methods
SNP selection
The T1D-associated SNP, rs1990760 (T946A), was selected to assess its effect onIFIH1expression levels. An intronic SNP was selected for each of the other 3 genes since there are no suitable common exonic SNPs. Intronic SNPs have been shown to yield
similar allelic expression levels to those obtained using exonic SNPs, provided that the genes are highly expressed in the tissue sample, and that the unspliced mRNA (or heteronuclear RNA [hnRNA]) can be successfully amplified and detected [26]. The three selected intronic SNPs were in high LD with each other and with the rs1990760 SNP (r2= 0.513–0.739) (Table 1), and had higher minor allele frequencies (MAF = 0.398–0.492) than the T1D-associated SNP (MAF = 0.392), in order to maximize the number of heterozygotes obtained.
Samples
Lymphoblastoid cell lines (LCLs) from the CEU collection were used. These were unrelated individuals residing in Utah with ancestry from western and northern Europe genotyped for millions of SNPs genome-wide, as part of the HapMap project. LCL samples heterozygous for all of the chosen SNPs were grown in RPMI 1640 medium (Gibco, CA, USA), supplemented with penicillin/streptomycin, 2mM L-glutamine, non-essential amino acids, and 15% heat-inactivated fetal bovine serum (Multicell, RI, USA). Cells were pelleted when they reached a density of about 1.06106cells/ml. Thymic and pancreatic samples were obtained from our collection of frozen fetal tissues as previously described [18,27]. Written informed consent was obtained from all individuals included in this study and was approved by the Research Ethics Board of the hospitals where the recruitments took place: for LCLs, under the auspices of the Centre de L’ ’e´tude du Polymorphisme Humain, Paris, France; for thymic and pancreatic samples, by the Royal Victoria Hospital Research Ethics Board (McGill University Health Centre), Montre´al, Que´bec, Canada.
DNA and RNA extraction
Extraction of nucleic acids from pancreatic and thymic tissues has been described elsewhere [28], and RNA integrity was assessed by the 2100 Bioanalyzer (Agilent, CA, USA). LCL genomic DNA was extracted using the QIAamp DNA Mini Kit (Qiagen, Germany) and RNA was isolated using the RNeasy Plus Mini Kit (Qiagen, Germany) following the manufacturer’s protocol.
cDNA synthesis and PCR
In a typical reaction, 2.5mg aliquots of total RNA were treated with 1 U of DNAse I for 30 minutes at 37uC following the manufacturer’s protocol (Ambion, TX, USA). Reverse transcrip-tion (RT) was carried out under standard conditranscrip-tions using random hexamer primers (Invitrogen, CA, USA) and 1000 ng of total unfragmented RNA, or RNA that has been subjected to chemical fragmentation according to the manufacturer’s protocol (Ambion, TX, USA), along with SuperScript II reverse transcriptase according to the manufacturer’s instructions (Invitrogen, CA, USA). RNA was also primed with oligo-dt primers. No detectable
Table 1.Pairwise Linkage Disequilibrium Coefficients of SNPs at theIFIH1 locus.
SNPs rs1990760 (IFIH1) rs7587426 (GCA) rs2075302 (FAP) rs2068330 (KCNH7)
rs1990760 (IFIH1) - 0.700 0.513 0.738
rs7587426 (GCA) 0.959 - 0.586 0.643
rs2075302 (FAP) 0.908 0.846 - 0.370
rs2068330 (KCNH7) 0.859 0.919 0.770
levels of RT-PCR product were observed in RNA samples if reverse transcriptase was omitted. PCR amplification for genomic DNA and cDNA samples for IFIH1, (along with a minus-RT control) was carried out using primers that amplify exon 15 and thus are capable of amplifying both DNA and cDNA. All other genes were studied using primers located in intronic regions, thus amplifying hnRNA and DNA. Primer and probe sequences are listed in Table S1. To ensure uniform conditions, each cDNA sample was assayed with its corresponding heterozygous genomic DNA.
Briefly, 40 ng of DNA or cDNA were combined with 10mM of each primer pair, 10 mM dNTPs, 50 mM MgCl2, 16 PCR Buffer, and 0.3 U ofTaqPolymerase (Invitrogen, CA, USA), in a total volume of 25mL. Each PCR reaction consisted of an initial denaturation step at 94uC for 5 min, followed by 35 cycles of denaturation at 94uC for 30 s; annealing at 49.5uC forIFIH1for 30 s and extension at 72uC for 30 s, as well as a final extension step of 7 min. GCA, FAP and KCNH7 were amplified using the same conditions as described above, with an annealing temper-ature of 56.5uC. PCR products were subjected to electrophoresis in a 1.5% agarose gel. All samples were run in parallel for each gene in each tissue type.
Single nucleotide primer extension (SNuPE)
In order to identify heterozygotes for the chosen marker polymorphisms, 38 thymic and 23 pancreatic samples were initially genotyped using single nucleotide primer extension with dideoxy-NTPs (dddideoxy-NTPs) labeled with different fluorochromes corresponding to each allele [23,29,30]. Briefly, the PCR amplicons were extracted and purified from agarose gels using columns from the QIAquick Gel Extraction Kit (Qiagen, Germany), following the manufactur-er’s protocol. Primer extension was then carried out by combining 2mL of the purified PCR product with 5mL of the ABI Prism SNaPshot Multiplex Kit (Applied Biosystems, CA, USA), 2.5mM of the appropriate extension probe, and 3mL of water, in a total volume of 10mL. Primer extension thermocycling conditions consisted of an initial step of 95uC for 2 minutes, followed by 25 cycles of 95uC for 10s, and 60uC for 35 s forIFIH1. For the other genes, the denaturation step of 95uC for 10s was followed by cycling at 50uC for 5s then 60uC for 30s. Following primer extension, the products were treated with 1 U of shrimp alkaline phosphatase (Roche, IN, USA) to remove unincorporated ddNTPs, for 1 hr at 37uC, and then the enzyme was deactivated for 15 min at 75uC. Aliquots of 1mL of SnaPshot product and 9mL of Hi-Di formamide were loaded into a 3100 DNA sequencer (Applied Biosystems, CA,USA). Products were electrophoresed on a 36-cm capillary array at 60uC. As with cDNA synthesis and PCR steps, all samples were run simultaneously for each gene in each tissue type. Data was processed using Genescan Analysis version 3.7 software (Applied Biosystems, CA,USA). Peak heights representing allele-specific extended primers were calculated using GeneScan in order to generate a ratio of allelic representation. The area under the curve of the peak representing a particular allele is proportional to the abundance of that allele in the sample. Once heterozygous samples were identified, they were re-run simultaneously with their corresponding cDNA to evaluate the relative abundance of the two alleles at a particular polymorphism, using the same protocol. Hence, this enabled us to use the PCR product of the same sequence from genomic DNA as a control for 1:1 stoichiometry, or a 50:50 allele ratio. For reproducibility purposes, all samples showing a relative allele difference greater than 40% than the genomic average would be retested two more times, in two separate RT-PCR reactions, along with the corresponding genomic DNA. This was not necessary, as all allele ratios fell within the 40% range.
Statistical Analysis
The ratio of one allele over the other for each SNP was calculated for the AI assay. The ratio for each sample was divided by the average genomic ratio for that assay batch in order to account for differences in probe and fluorochrome efficiencies. Peak height ratios corre-sponding to each allele in individual DNA samples ranged from 0.79 to 1.21 forIFIH1, from 0.80 to 1.35 forGCA, and from 0.93 to 1.06 forFAPSNP. Allelic expression differences between DNA and cDNA were evaluated by the student t test for statistical significance. A two-tailed level of 0.05 was chosen for a type I error rate. Power analysis was calculated for detection of a 40% difference in relative expression of the two alleles, ata= 0.01 (similar to Liuet al.[14]).
Results
Our working sample was comprised of 9 LCL, 13 thymus, and 6 pancreas samples that were found to be heterozygous for all four SNPs.IFIH1 andGCAgene expression was detected in all LCL, thymus and pancreas samples.FAP was exclusively expressed in pancreas and thymus, but not LCLs. We were not able to detect
KCNH7expression in any of the three tissues.
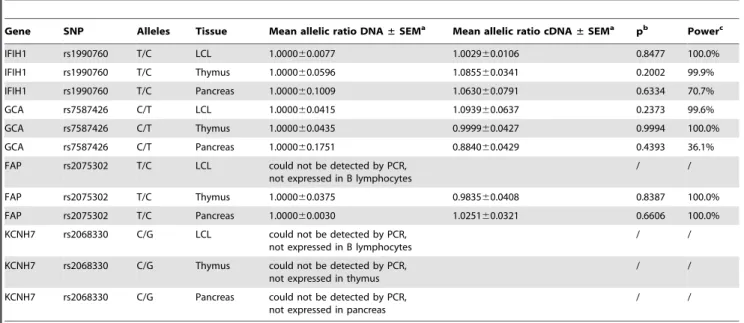
The calculated allelic ratios for each SNP representing each gene in theIFIH1locus and their distribution in the different tissues that were assayed are shown in Table 2 and Figure 1 respectively. The allelic ratio distribution ofIFIH1, GCA, andFAP cDNA do not significantly differ from that of their corresponding DNA. After correction by the genomic DNA allele proportion, the average ratio (mean6SEM) of the major allele (T) over the minor allele (C) of the rs1990760 SNP inIFIH1in LCLs is 1.002960.0106, p= 0.8477 (Table 2), indicating the absence of an AI effect due to a common genetic variation at theIFIH1gene in LCLs. The same is observed in pancreas and thymus. No evidence of significant transcriptional effect was seen in any of the other genes in all the assayed tissues and cells (Table 2).Our approach had a 99% power to detect a transcriptional effect of rs1990760 on IFIH1 in LCLs, of the magnitude reported by Liu et al. [14] (40% allelic difference at a= 0.01). We also have 99% power to detect a 25% difference in expression between the twoIFIH1alleles. Since all RNA samples were DNAse-treated prior to RT-PCR and did not generate detectable PCR product in the absence of an RT step, it is highly improbable that our results were influenced in some way by genomic DNA contamination. It has been recently suggested [31] that differential secondary structure of RNA alleles may interfere with quantitative comparisons through a differential effect on the efficiency of reverse transcription, creating spurious allelic imbal-ance or conceivably masking true imbalimbal-ance (if it happens to be exactly equal and in the opposite direction). To deal with this, minimization of secondary structure by fragmenting the RNA prior to reverse transcription (RT) was recommended. To see whether this may be a problem in the specific case ofIFIH1, we compared allelic ratios obtained with or without fragmentation of the RNA prior to RT. In twelve independent comparisons, the mean allelic ratio (normalized for the average DNA ratio) was 0.9560.07 (SEM) for unfragmented vs. 1.0060.05 for fragmented RNA (p = 0.56, 99% power to detect a 40% effect ata= 0.01, 86.7% power to detect a 25% effect at a= 0.05). Oligo-dT priming of the RT, suggested as an alternative, also gave nearly-identical results (0.9860.05 [SEM]). We, therefore, concluded that interference by secondary structure was not an issue in allelicIFIH1measurements. Discussion
Table 2.Summary of difference in allelic variation at theIFIH1locus.
Gene SNP Alleles Tissue Mean allelic ratio DNA±SEMa Mean allelic ratio cDNA
±SEMa pb Powerc
IFIH1 rs1990760 T/C LCL 1.000060.0077 1.002960.0106 0.8477 100.0%
IFIH1 rs1990760 T/C Thymus 1.000060.0596 1.085560.0341 0.2002 99.9%
IFIH1 rs1990760 T/C Pancreas 1.000060.1009 1.063060.0791 0.6334 70.7%
GCA rs7587426 C/T LCL 1.000060.0415 1.093960.0637 0.2373 99.6%
GCA rs7587426 C/T Thymus 1.000060.0435 0.999960.0427 0.9994 100.0%
GCA rs7587426 C/T Pancreas 1.000060.1751 0.884060.0429 0.4393 36.1%
FAP rs2075302 T/C LCL could not be detected by PCR, not expressed in B lymphocytes
/ /
FAP rs2075302 T/C Thymus 1.000060.0375 0.983560.0408 0.8387 100.0%
FAP rs2075302 T/C Pancreas 1.000060.0030 1.025160.0321 0.6606 100.0%
KCNH7 rs2068330 C/G LCL could not be detected by PCR, not expressed in B lymphocytes
/ /
KCNH7 rs2068330 C/G Thymus could not be detected by PCR, not expressed in thymus
/ /
KCNH7 rs2068330 C/G Pancreas could not be detected by PCR, not expressed in pancreas
/ /
an = 9 for LCLs, n = 13 for thymus, n = 6 for pancreas.
bstatistical significance as measured by the two-tailed studentttest.
cstatistical power to detect a 40% difference of expression between alleles, at ana= 0.01. doi:10.1371/journal.pone.0011564.t002
Figure 1. Allelic ratio distribution at theIFIH1locus.9 Lymphoblastoid cell lines (LCL), 6 pancreas (Panc.) and 13 thymus (Thym.) tissue from individuals heterozygous for the selected marker SNPs for each gene were used to assess allelic imbalance at theIFIH1 locus. Relative allelic
abundance in individual samples has been normalized to the mean genomic DNA ratio (equal to1) and normalized sample RNA ratios were compared to those of normalized genomic DNA for each gene in each tissue. The average means6SEM are summarized in table 2, along with the statistical analysis. Our power to detect a difference of 40% in the means of DNA and LCL RNA was.99%.
strong LD in the block has made it far more challenging to identify the disease-causing polymorphism or haplotype. Thus, detailed functional analysis is required. A recent study that showed higher
IFIH1mRNA levels in PBMCs of individuals with the susceptible genotype of the T1D associated SNP by real-time RT-PCR suggested that a differential regulation ofIFIH1expression could be, at least in part, responsible for the T1D risk [14], while another study was not able to show any difference in IFIH1 allelic expression using the same technology [16]. This approach, comparingIFIH1levels across individuals with different genotypes [14,16], introduces substantial noise from inter-individual vari-ability and by factors such as assay and batch variance,trans-acting genetic factors, immune experience, cell proportions in the PBMC mixtures and the presence of polymorphisms in other genes that may alter the expression level in trans[23,34], thus diluting the effect ofcis-acting influences in expression studies [35]. In order to resolve this controversy, we used SNuPE, a method that relies on the comparison of alleles within rather than between samples, which removes external confounding factors. The SNuPE technique is quite accurate and thus allows small differences in allelic ratios to be reliably detected [24]. We have previously validated the accuracy of this method by showing an excellent correlation between observed and expected allelic ratios [19].
The concern that allelic expression in transformed cultured cell lines may not accurately represent what occurs in human tissues was addressed in a recent paper that explored whether different culture conditions (passage number, pH, nutrient concentration, cell density, etc.) influence AI results [26]. It was found that estimations of AI after different passages were not significantly different from one another. Another potential limitation of our study was our use of total pancreas rather than islets. Since AI of some genes may be tissue specific, we may have missed an islet-specific effect. This, however, seems unlikely since both endocrine and exocrine pancreatic tissues are of similar origins, and likely exhibit similar allelic expression.
We were unable to replicate the results reported by Liuet al.
[14], using a much more sensitive, accurate and reproducible method that has the power to detect an effect that is similar to that was reported. This is in accordance with the expectation that the nsSNP is the most likely functional candidate.
In summary, our results suggest that GCA and FAP are not involved in T1D since we observed no AI and there are no nsSNPs of high enough frequency to explain the effect [1]. This reinforces the role of the IFIH1 nsSNP as a potential causal variant. In addition, KCNH7 was not expressed in LCLs, fetal pancreas or thymus, and thus could not be assayed for AI. Therefore, a transcriptional effect of the Thr946Ala SNP, or any variant in LD with it, onKCNH7cannot be ruled out.
While our study shows no transcriptional effects of the Thr946Ala SNP or the other chosen variants that were in tight LD with it on the three assayed genes, we cannot exclude the possibility of the presence of othercis-regulatory variants with a lower minor allele frequency exerting transcriptional effects on these genes, and being detected by our AI assay. While in some samples, the cDNA allelic ratio deviates by at least 20% from the genomic ratio (Figure 1), we are unable to conclude whether we are observing transcriptional effects of such a polymorphism incis, or whether these merely reflect measurement error. Nonetheless, theIFIH1gene has been sequenced extensively,
and no rare variants have been found that can explain the association of the common Thr946Ala SNP to T1D [8].
Thus, the mechanism of the observed association of the rs1990760 with T1D remains to be determined, but does not involve modulation of mRNA. Although the IFIH1 nsSNP, rs1990760 (Thr946Ala substitution), does not reside in either the signaling CARD domain or RNA binding helicase domain of the protein, this region of the protein is conserved between mammals and may have other, unknown functions or have an effect on the active domains through changes in tertiary structure [1], which may well be affected by the Thr946Ala SNP. This is a non-conservative substitution, changing the polarity of the amino acid from polar to non-polar. Recently, four rare mutations have been identified in
IFIH1, each of which separately lowered the risk of developing T1D [8], independently of the effect of the Thr946Ala SNP. Two of these four variants have been shown to be loss of function mutations, dramatically reducingIFIH1protein activity or its RNA binding ability [9]. That loss ofIFIH1function protects from T1D would indicate that the risk allele is related to an exaggerated immune response rather than imperfect anti-viral defense. By implication, if Thr946Ala itself is indeed the functional variant responsible for its T1D association, one would expect the diabetes-predisposing Ala allele to represent gain of function even though it was derived by mutation of the conserved ancestral Thr allele. However, such a mutation needs not to result in diminished protein function, as conservation is driven by fitness of the organism, not higher level of protein function. Such an example can be found in one of the strongest T1D associations, which involves the R620W SNP in
PTPN22 (protein tyrosine phosphatase, non-receptor type 22), where the 620W disease-associated variant, derived in an even more conserved context, is a gain-of-function variant, with increased catalytic activity [36]. In the same paper which showed that two of the four rareIFIH1 variants are loss of function mutations, the Thr946Ala nsSNP was not found to affect dsRNA binding or consequent IFN gene activation in mouse embryonic fibroblast cells in culture [9]. However, in a transfection system, an excess ofIFIH1, expressed above the threshold where small allelic differences in dsRNA binding affinity and/or IFN response can be detected, subtle effects could have been missed. It may also be that the SNP alters interaction with specific dsRNA structures not modeled by the mimic used. Alternatively, the Thr946Ala nsSNP could affect translational efficiency or may even be involved in the post-translational processing of theIFIH1protein. Additional work on the Thr946Ala SNP is therefore necessary to discover how it alters
IFIH1function and gain insight on how it affects T1D pathogenesis. Supporting Information
Table S1 Primer sequences and probes for each SNP
Found at: doi:10.1371/journal.pone.0011564.s001 (0.02 MB XLS)
Author Contributions
Conceived and designed the experiments: HZ CP. Performed the experiments: HZ. Analyzed the data: HZ CP. Contributed reagents/ materials/analysis tools: LM CP. Wrote the paper: HZ CP. Provided technical expertise with experiments and interpretation of data, helped revise the manuscript critically: LM.
References
1. Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, et al. (2006) A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet 38: 617–619.
3. Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, et al. (2002) mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci U S A 99: 637–642.
4. Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, et al. (2005) Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol 175: 2851–2858. 5. Hyoty H, Taylor KW (2002) The role of viruses in human diabetes.
Diabetologia 45: 1353–1361.
6. Roivainen M (2006) Enteroviruses: new findings on the role of enteroviruses in type 1 diabetes. Int J Biochem Cell Biol 38: 721–725.
7. van der Werf N, Kroese FG, Rozing J, Hillebrands JL (2007) Viral infections as potential triggers of type 1 diabetes. Diabetes Metab Res Rev 23: 169–183. 8. Nejentsev S, Walker N, Riches D, Egholm M, Todd JA (2009) Rare variants of
IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 324: 387–389.
9. Shigemoto T, Kageyama M, Hirai R, Zheng J, Yoneyama M, et al. (2009) Identification of loss of function mutations in human genes encoding RIG-I and MDA5: implications for resistance to type I diabetes. J Biol Chem 284: 13348–13354.
10. Goldstein DB (2009) Common genetic variation and human traits. N Engl J Med 360: 1696–1698.
11. Boyhan A, Casimir CM, French JK, Teahan CG, Segal AW (1992) Molecular cloning and characterization of grancalcin, a novel EF-hand calcium-binding protein abundant in neutrophils and monocytes. J Biol Chem 267: 2928–2933. 12. Fassnacht M, Lee J, Milazzo C, Boczkowski D, Su Z, et al. (2005) Induction of CD4(+) and CD8(+) T-cell responses to the human stromal antigen, fibroblast activation protein: implication for cancer immunotherapy. Clin Cancer Res 11: 5566–5571.
13. Muhlbauer E, Bazwinsky I, Wolgast S, Klemenz A, Peschke E (2007) Circadian changes of ether-a-go-go-related-gene (Erg) potassium channel transcripts in the rat pancreas and beta-cell. Cell Mol Life Sci 64: 768–780.
14. Liu S, Wang H, Jin Y, Podolsky R, Reddy MV, et al. (2009) IFIH1 polymorphisms are significantly associated with type 1 diabetes and IFIH1 gene expression in peripheral blood mononuclear cells. Hum Mol Genet 18: 358–365.
15. Dixon AL, Liang L, Moffatt MF, Chen W, Heath S, et al. (2007) A genome-wide association study of global gene expression. Nat Genet 39: 1202–1207. 16. Marinou I, Montgomery DS, Dickson MC, Binks MH, Moore DJ, et al. (2007)
The interferon induced with helicase domain 1 A946T polymorphism is not associated with rheumatoid arthritis. Arthritis Res Ther 9: R40.
17. Yan H, Yuan W, Velculescu VE, Vogelstein B, Kinzler KW (2002) Allelic variation in human gene expression. Science 297: 1143.
18. Vafiadis P, Bennett ST, Todd JA, Nadeau J, Grabs R, et al. (1997) Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat Genet 15: 289–292.
19. Anjos SM, Shao W, Marchand L, Polychronakos C (2005) Allelic effects on gene regulation at the autoimmunity-predisposing CTLA4 locus: a re-evaluation of the 39+6230G.A polymorphism. Genes Immun 6: 305–311.
20. Pastinen T, Hudson TJ (2004) Cis-acting regulatory variation in the human genome. Science 306: 647–650.
21. Pastinen T, Sladek R, Gurd S, Sammak A, Ge B, et al. (2004) A survey of genetic and epigenetic variation affecting human gene expression. Physiol Genomics 16: 184–193.
22. Singer-Sam J, LeBon JM, Dai A, Riggs AD (1992) A sensitive, quantitative assay for measurement of allele-specific transcripts differing by a single nucleotide. PCR Methods Appl 1: 160–163.
23. Bray NJ, O’Donovan MC (2006) Investigating cis-acting regulatory variation using assays of relative allelic expression. Psychiatr Genet 16: 173–177. 24. Buckland PR (2004) Allele-specific gene expression differences in humans. Hum
Mol Genet 13 Spec No 2: R255–R260.
25. Singer-Sam J, Chapman V, LeBon JM, Riggs AD (1992) Parental imprinting studied by allele-specific primer extension after PCR: paternal X chromosome-linked genes are transcribed prior to preferential paternal X chromosome inactivation. Proc Natl Acad Sci U S A 89: 10469–10473.
26. Serre D, Gurd S, Ge B, Sladek R, Sinnett D, et al. (2008) Differential allelic expression in the human genome: a robust approach to identify genetic and epigenetic cis-acting mechanisms regulating gene expression. PLoS Genet 4: e1000006.
27. Marchand L, Polychronakos C (2007) Evaluation of polymorphic splicing in the mechanism of the association of the insulin gene with diabetes. Diabetes 56: 709–713.
28. McCann JA, Zheng H, Islam A, Goodyer CG, Polychronakos C (2001) Evidence against GRB10 as the gene responsible for Silver-Russell syndrome. Biochem Biophys Res Commun 286: 943–948.
29. Hoogendoorn B, Norton N, Kirov G, Williams N, Hamshere ML, et al. (2000) Cheap, accurate and rapid allele frequency estimation of single nucleotide polymorphisms by primer extension and DHPLC in DNA pools. Hum Genet 107: 488–493.
30. Norton N, Williams NM, Williams HJ, Spurlock G, Kirov G, et al. (2002) Universal, robust, highly quantitative SNP allele frequency measurement in DNA pools. Hum Genet 110: 471–478.
31. Dendrou CA, Plagnol V, Fung E, Yang JH, Downes K, et al. (2009) Cell-specific protein phenotypes for the autoimmune locus IL2RA using a genotype-selectable human bioresource. Nat Genet 41: 1011–1015.
32. Qu HQ, Marchand L, Grabs R, Polychronakos C (2008) The association between the IFIH1 locus and type 1 diabetes. Diabetologia 51: 473–475. 33. Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, et al. (2007) Robust
associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet 39: 857–864.
34. Bray NJ, Buckland PR, Owen MJ, O’Donovan MC (2003) Cis-acting variation in the expression of a high proportion of genes in human brain. Hum Genet 113: 149–153.
35. Pastinen T, Ge B, Gurd S, Gaudin T, Dore C, et al. (2005) Mapping common regulatory variants to human haplotypes. Hum Mol Genet 14: 3963–3971. 36. Vang T, Congia M, Macis MD, Musumeci L, Orru V, et al. (2005)