J. Braz. Chem. Soc. vol.21 número8
Texto
Imagem
Documentos relacionados
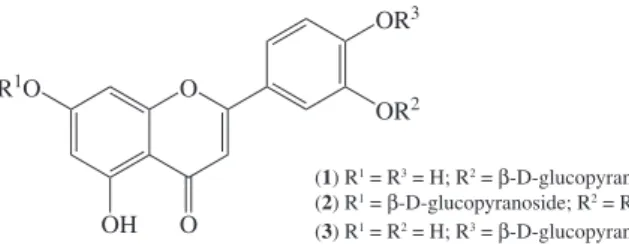
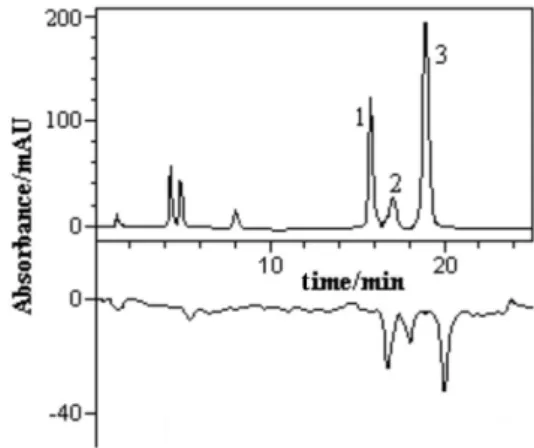
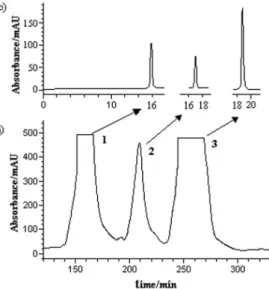
The composition of the essential oil of Eugenia dysenterica collected from wild populations in Senador Canedo (SC) and Campo Alegre de Goiás (CA), as well as from
Table 4 shows the concentrations and standard deviations for the heavy metals in organic residue samples collected in ive coffee processing plants located around of
This result might also indicate that both series of compounds have higher antioxidant action in a mechanism different of electron scavenging. For the two methods tested, an
A miniaturized enzymatic assay using luorescent probes to reveal esterase producing microorganisms was optimized and applied to screen 64 soil bacterial strains.. The
Results suggest that in acid pH weak electrostatic interactions and hydrogen bonding are responsible for aggregates formation while in alkaline pH electrostatic interactions
where F o is DPH luorescence intensity in organic solvent, F is observed luorescence intensity, K SV is Stern-Volmer quenching constant and [H 2 O] is water molar concentration.
The results here presented support a random order kinetic mechanism of addition of substrates, and indicate that DD-CoA binding to free WT InhA follows the sequential model,
Herein, we report our current investigations in the study of the Candida tropicalis CE017 strain behavior as a novel stereoselective reducing agent of aromatic
