UFOP - CETEC - UEMG
REDEMAT
R
EDET
EMÁTICA EME
NGENHARIA DEM
ATERIAISUFOP – CETEC – UEMG
Dissertação de Mestrado
"Síntese e caracterização de géis de acetato de
celulose reticulados com dianidrido piromelítico e
dianidrido do ácido 3,3´,4,4´benzofenona
tetracarboxílico"
Autor: Víctor de Alvarenga Oliveira
Orientador: Prof. Vagner Roberto Botaro
Co-orientador: Cláudio G. dos Santos
UFOP - CETEC - UEMG
REDEMAT
R
EDET
EMÁTICA EME
NGENHARIA DEM
ATERIAISUFOP – CETEC – UEMG
Víctor de Alvarenga Oliveira
"Síntese e caracterização de géis de acetato de celulose
reticulados com dianidrido piromelítico e dianidrido do ácido
3,3´,4,4´benzofenona tetracarboxílico"
Dissertação de Mestrado apresentada ao Programa de Pós-Graduação em Engenharia de Materiais da REDEMAT, como parte integrante dos requisitos para a obtenção do título de Mestre em Engenharia de Materiais.
Área de concentração: Análise e seleção de materiais
Orientador: Prof. Vagner Roberto Botaro
Co-orientador: Cláudio G. dos Santos
Catalogação: [email protected] O48s Oliveira, Victor de Alvarenga.
Síntese e caracterização de géis de acetato de celulose reticulados com
dianidrido piromelítico e dianidrido do ácido 3,3’,4,4’ benzofenona tetracarboxílico [manuscrito] / Victor de Alvarenga Oliveira. – 2008.
xv, 137f.: il. color., grafs e tabs.
Orientador: Prof. Dr. Vagner Roberto Botaro.
Dissertação (Mestrado) - Universidade Federal de Ouro Preto. Escola de Minas. Rede Temática em Engenharia de Materiais.
Área de concentração: Análise e seleção de materiais.
1. Acetato de celulose - Teses. 2. Acetatos - Teses. 3. Hidrogel - Teses. I. Universidade Federal de Ouro Preto. Escola de Minas. II. Título.
Agradecimentos
À minha mãe por ser grande e assim fazer todas as conquistas parecerem pequenas. Agradeço à Lorena pela dedicação, amor e paciência.
Aos meus irmãos que estão sempre presentes em minhas conquistas. Ao meu pai pelo apoio e confiança.
Agradeço ao Maurílio pela paciência e sabedoria.
Agradeço aos meus tios em especial ao Tio Inácio pela ajuda de sempre e ao Tibeto por ser companheiro. Às minhas tias Glória, Vera e em especial à Tia Miriam.
Agradeço aos meus primos André, Alexandre Folhão e Henrique. À Daniela Preá pela ajuda e à Bela (pelo dente).
Agradeço ao meu orientador Vagner por acreditar.
Abstract
This work describes the synthesis of hydrogels of cellulose acetate (AC), with a nominal degree of substitution DS = 2.5,
cross-linked with 1,2,4,5-benzenotetracarboxylic dianhydride (PMDA) and 3,3',4,4'-benzophenonetetracarboxylic dianhydride (BTDA).
Hydrogels were obtained by esterification of the free hydroxyl groups in AC, using triethylamine as catalyst and acetone as solvent.
The raw materials were characterized by thermal analysis (TG/DTG - DSC - DTA) and by Fourier transform infrared spectroscopy (FTIR). DS of cellulose acetate was determined by titration with a known amount of standard NaOH solution.
Hydrogels of PMDA were synthesized in stoichiometric ratios 0.1, 0.25, 0.5, 0.75 and 1.0 mol of PMDA/mol of AC and hydrogels of BTDA were synthesized instoichiometric ratios 0.5, 0.75 and 1.0 mol of
BTDA/mol of AC.
FTIR proved to be a suitable method to follow the course of reactions and the progress of purification. UV-vis spectroscopy and thermal analysis confirmed the esterification of the free hydroxyl groups. Surface modification of AC structure after the cross-linking reaction was analyzed by Scanning Electron Microscope (SEM). Density and porosity of the hydrogels were determined by BET.
The influence of the concentration of dianhydride on the time necessary for formation of the gel was investigated. The influence of increasing in the degree of cross-linking on the thermal behavior of the material was also documented.
long durations of experiments and the third described the mechanism as a whole.
The Arrhenius equation was used to determine the diffusion coefficient of the different hydrogels in different temperatures and the threshold energy for the swelling process. The swelling kinetic of gels was analyzed using the second order equation of Schott.
The enthalpy of mixture ∆Hmix of the gel-water system was
Resumo
Nesse trabalho foram realizadas sínteses de hidrogéis de acetato de celulose (AC), com grau de substituição 2,5, reticulados com dianidrido piromelítico (PMDA) e dianidrido da 3, 3´, 4, 4´ benzofenona tetracarboxílico (BTDA), por intermédio da esterificação das hidroxilas livres nas cadeias de AC.
As caracterizações das matérias primas foram realizadas por análises térmicas (TG/DTG – DSC – DTA) e espectroscopia na região do infravermelho (FTIR). O grau de substituição do acetato de celulose (AC) foi determinado por via úmida.
Hidrogéis de PMDA foram sintetizados com razões estequiométricas de 0,1, 0,25, 0,5, 0,75 e 1,0 mol de PMDA/ mol de AC e hidrogéis de BTDA foram
sintetizados com razões estequiométricas de 0,5, 0,75 e 1,0 mol de BTDA/ mol de AC. A confirmação da reação de esterificação foi possível a partir de análises de técnicas de espectroscopia na região do infravermellho (FTIR), espectroscopia na região do ultravioleta (UV-vis) e análises térmicas (TG/DTG – DSC – DTA). Modificações superficiais e a presença de poros no novo material foram analisadas através de microscopia eletrônica de varredura (MEV) e porosimetria (BET), respectivamente.
A influência da concentração de dianidrido, no meio reacional, no tempo de formação do gel foi investigada, bem como a influência do aumento no grau de reticulação no comportamento térmico do material.
Isotermas de absorção de água foram obtidas para os hidrogéis com diferentes agentes reticuladores e diferentes graus de reticulação em diferentes temperaturas. A análise do mecanismo de inchamento dos géis foi feita em três etapas: a primeira etapa analisa os primeiros 60% da isoterma de inchamento, a segunda etapa descreve o mecanismo de relaxação das cadeias para grandes tempos de experimento (fração superior a 60%) e a terceira etapa descreve o mecanismo como um todo.
O coeficiente de difusão dos diferentes géis em diferentes temperaturas foi determinado, a energia de ativação para o processo de inchamento foi determinada, utilizando a equação de Arrhenius, e a cinética de inchamento dos géis foi analisada utilizando a equação de segunda ordem de Schott.
A entalpia de mistura ∆Hmix do sistema gel-água pôde ser determinada através da
Sumário
Capítulo 1
1
1. Introdução 1
1.1. A utilização da celulose como matéria prima 1
1.2. A produção de celulose no Brasil 1
1.3. O acetato de celulose como matéria prima 2
1.4. A produção de acetato de celulose no Brasil 3
1.5. Hidrogéis de acetato de celulose 4
1.6. Objetivos 7
1.6.1. Gerais 7
1.6.2. Específicos 7
Capítulo 2
8
2. Revisão bibliográfica 8
2.1. Celulose 8
2.2. Estruturas cristalinas da celulose 9
2.3. As regiões amorfas da celulose 11
2.4. Ésteres de Celulose 13
2.5. Acetato de Celulose 14
2.6. Redes poliméricas 16
2.7. Hidrogéis 17
2.7.1. Hidrogéis neutros 20
2.7.2. Hidrogéis iônicos 22
2.8. Processos de absorção de água pelos hidrogéis 24
2.8.1. Primeira lei da difusão 24
2.8.2. Segunda lei de Fick 25
2.8.3. O processo de inchamento em hidrogéis 26
2.8.4. Análise da dinâmica de inchamento de hidrogéis 27
2.9. Agente reticulador 31
2.9.1. Dianidridos 31
Capítulo 3
34
3. Materiais e Métodos 34
3.1. Síntese dos géis 34
3.1.1. Matéria Prima 34
3.1.2. Purificação dos reagentes 34
3.1.3. Determinação do ponto de fusão da matéria prima 35
3.1.4. Análise Térmica Diferencial (DTA) 35
3.1.5. Determinação do grau de acetilação do Acetol Flakes® 35
3.1.6. Síntese dos géis de PMDA e BTDA 36
3.1.7. Processo de lavagem e secagem 37
3.2. Estudo cinético da reação 37
3.3. Espectroscopia de absorçãono infravermelho (FTIR) 38 3.4. Termogravimetria (TG) e termogravimetria derivada (DTG) 38
3.5. Calorimetria Exploratóri Diferencial (DSC) 39
3.6. Microscopia Eletrônica de Varredura (MEV) 39
3.7. Adsorção de Nitrogênio (BET) e densidade 39
3.8. Espectroscopias de absorção no ultravioleta (UV-vis) 39
3.9. Estudo do inchamento 40
Capítulo 4
41
4. Apresentação e discussão dos resultados 41
4.1. Resultados da síntese dos géis 41
4.1.1. Resultado de caracterização das matérias-primas 41
4.1.1.a. Pontos de fusão 41
4.1.1.b. Espectroscopia na região do infravermelho 41
4.1.1.c. Análises térmicas 44
4.1.2. Apresentação dos resultados de análise do grau de substituição do
Acetol Flakes® 53
4.1.3. Discussão dos resultados de síntese, secagem e lavagem dos géis 54
4.2. Apresentação e discussão dos resultados cinéticos 59 4.3. Apresentação e discussão dos resultados de TG e DTG 64
4.4. Resultados de DSC 67
4.5. Resultados de adsorção de Nitrogênio (BET) 68
4.6. Resultados de Microscopia Eletrônica de Varredura (MEV) 68
4.7. Análise qualitativa do grau de esterificação 69
4.8. Resultados de inchamento 72
4.8.1 (a). Apresentação dos resultados de inchamento para os géis de PMDA 73
4.8.1 (b). Apresentação dos resultados de inchamento para os géis de BTDA 76
4.8.1 (c). Comparação dos resultados de absorção de água para os géis de
PMDA e BTDA 80
4.8.2. Mecanismo de inchamento 81
4.8.2.1. Mecanismo de inchamento para frações de água absorvida
inferiores a 0,6 81
4.8.2.1 (a). Mecanismo de inchamento da fração de água absorvida inferior
a 0,6, para os géis de PMDA 82
4.8.2.1 (b). Mecanismo de inchamento da fração de água absorvida inferior
a 0,6, para os géis de BTDA 85
4.8.2.1 (c). Mecanismo de inchamento da fração de água absorvida inferior
a 0,6, para os géis de PMDA e BTDA 87
4.8.2.2. Análise da fração de água absorvida superior a 0,6 88
4.8.2.2 (a). Análise da fração de água absorvida superior a 0,6, para os géis
de PMDA 88
4.8.2.2 (b). Análise da fração superior a 0,6 dos géis de BTDA 92 4.8.2.2 (c). Análise da fração superior a 0,6 dos géis de PMDA e BTDA 95
4.8.2.3. Análise do mecanismo de absorção para toda a isoterma de
inchamento 96
4.8.2.3 (a). Análise do mecanismo de absorção para toda a isoterma de
inchamento, para os géis de PMDA 96
4.8.2.3 (b). Análise do mecanismo de absorção para toda a isoterma de
inchamento, para os géis de BTDA 103
4.8.2.3 (c). Análise do mecanismo de absorção para toda a isoterma de
inchamento, para os géis de PMDA e BTDA 108
4.8.3. A energia de ativação do processo de inchamento 108
4.8.3 (a). A energia de ativação do processo de inchamento, para os géis de
PMDA 108
4.8.3 (b). A energia de ativação do processo de inchamento, para os géis de
BTDA 109
4.8.3 (c). A energia de ativação do processo de inchamento, para os géis de
PMDA e BTDA 110
4.8.4. Cinética de inchamento 111
4.8.5. A entalpia de mistura do sistema gel-água 118
4.6.5 (a). A entalpia de mistura do sistema gel-água para os géis de PMDA 118
4.8.5 (b). A entalpia de mistura do sistema gel-água para os géis de BTDA 122
4.8.5 (c). A entalpia de mistura do sistema gel-água para os géis de PMDA e BTDA 123
4.9. Descrição de um mecanismo simplificado e possível para a reação 123
Capítulo 5
129
5. Conclusões 129
Capítulo 6
132
Lista de Figuras
Figura 1.1. - Fotografia do Acetol Flakes→ 2
Figura 1.2. - Estrutura química do hidrogel de PMDA 4
Figura 1.3. - Estrutura química do hidrogel de BTDA. 5
Figura 1.4. - Hidrogéis de acetato de celulose reticulados com PMDA. Na figura A mostramos o gel 0,1P com ampliação de 8X, na figura B mostramos o gel 1,0P com uma ampliação de10X, na figura C mostramos o gel 0,25P com ampliação de 50X e na figura D mostramos o gel 0,25P com ampliação de 6,5X. 5
Figura 1.5. - Hidrogéis de acetato de celulose reticulados com BTDA. Na figura A mostramos o gel 0,5B com ampliação de 6,5X, na figura B mostramos o gel 0,5B com ampliação de 8X e na figura C mostramos o gel 3,0B com ampliação de 50X. 5
Figura 2.1.1. - Unidade de (β) D-glucopiranose. 8
Figura 2.1.2. - A estrutura circulada pela linha pontilhada representa a unidade de anidrocelobiose. 9
Figura 2.2.1. - Algumas das possíveis ligações de hidrogênio intra e intermolecular formadas entre as cadeias de celulose. 11
Figura 2.5.1. - Estrutura química do acetato de celulose. 15
Figura 2.6.1. - Formação de uma rede polimérica através da polimerização de grupos terminais. 16
Figura 2.6.2. - Formação de uma rede polimérica através da reticulação das cadeias. 17 Figura 2.9.1.1. - Estrutura química do dianidrido do ácido 1,2,4,5 benzeno tetracarboxílico (PMDA). 31
Figura 2.9.1.2. - Estrutura química do dianidrido do ácido 3,3´,4,4´-benzofenona tetracarboxílico (BTDA). 31
Figura 2.9.2.1. - Ligação intramolecular presente em poli ácidos carboxílicos. 33
Figura 4.1.1. - Espectro de FTIR do acetato de celulose (Acetol Flakes®). 42
Figura 4.1.2. - Espectro de FTIR do acetato de celulose padrão. 42
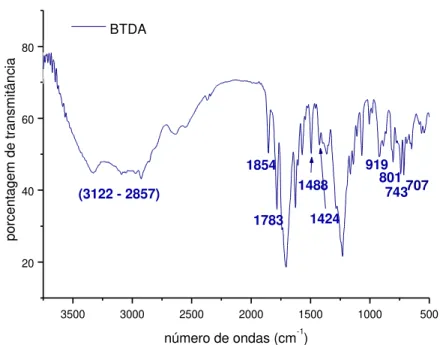
Figura 4.1.3. - Espectro de FTIR do BTDA. 43
Figura 4.1.5. - A - curva termogravimétrica (TG), B - curva termogravimétrica derivada
(DTG) e C - calorimetria exploratória diferencial (DSC) do AC. 45
Figura 4.1.6. - A - curva termogravimétrica (TG), B - curva termogravimétrica derivada
(DTG) e C - curva térmica diferencial (DTA) do AVICEL. 47
Figura 4.1.7. - A - Curva termogravimétrica (TG), B - curva termogravimétrica
derivada (DTG) e C - curva térmica diferencial (DTA) do BTDA. 49
Figura 4.1.8. - Reação atribuída ao primeiro evento da curva de DTG do BTDA. 49
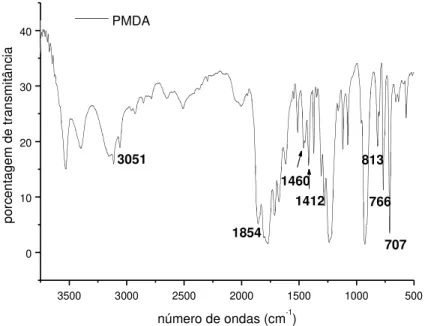
Figura 4.1.9. - Curva termogravimétrica (TG), B - curva termogravimétrica derivada
(DTG) e C - curva térmica diferencial (DTA) do PMDA. 51
Figura 4.1.10. - Reação atribuída ao segundo evento da curva de DTG do PMDA. 51
Figura 4.1.3.1. - FTIR do acetato de celulose (a), do gel de PMDA lavado (b) e do gel
de PMDA não lavado (c). 56
Figura 4.1.3.1. - FTIR do acetato de celulose (a), do gel de BTDA lavado (b) e do gel
de BTDA não lavado(c). 58
Figura 4.1.3.3. - TG do acetato de celulose e dos géis de BTDA e PMDA. 59
Figura 4.1.3.4. - DTG do acetato de celulose e dos géis de BTDA e PMDA. 59
Figura 4.2.1. - Curva cinética mostrando a influência da concentração inicial de
dianidrido no tempo gasto para o sistema alcançar o ponto de gel. 60
Figura 4.2.2. - Curvas que mostram o decaimento de primeira ordem quando as
concentrações de dianidridos foram menores ou pouco maiores que o ponto de formação
dos precipitados. 61
Figura. 4.2.3. - Formas canônicas no grupo carbonila de uma cetona. 62
Figura 4.2.4. - Formas canônicas de grupos anidridos. 62
Figura 4.2.5. - A molécula de PMDA e as posições ativadas por cada grupos ligado ao
anel. 63
Figura 4.2.6. - A molécula de BTDA e as posições ativadas por cada grupos ligado ao
anel. 63
Figura 4.3.1. - DTG e TG dos géis de BTDA com diferentes razões estequiométricas.
64
Figura 4.3.2. - DTG e TG dos géis de PMDA com diferentes razões estequiométricas.
65
Figura 4.3.3. - Desidratação dos dianidridos que reagiram com apenas uma cadeia de
Figura 4.4.1. - Curvas de DSC para os geís de BTDA. 67
Figura 4.4.2. - Curvas de DSC para os géis de PMDA. 68
Figura 4.6.1. - Microfotografia da superfície do AC. 69
Figura 4.6.2. - Microfotografia da superfície do gel 0.5 de PMDA. 69
Figura 4.6.3. - Microfotografia da superfície do gel 0.5 de BTDA. 69
Figura 4.7.1. - FTIR dos géis de PMDA com diferentes razões estequiométricas após desesterificação e do acetato de celulose. 70
Figura 4.7.2. - FTIR dos géis de BTDA com diferentes razões estequiométricas após desesterificação e do acetato de celulose. 71
Figura 4.7.3. - Espectro na região do ultravioleta para os fragmentos do gel de BTDA. 71
Figura 4.7.4. - Espectro na região do ultravioleta para os fragmentos do gel de PMDA. 72
Figura (a) 4.8.1.1. - Isotermas de inchamento para os géis de PMDA com diferentes razões estequiométricas, a 20ºC. 74
Figura (a) 4.8.1.2. - Isotermas de inchamento para os géis de PMDA, com diferentes razões estequiométricas, a 30ºC. 75
Figura (a) 4.8.1.3. - Isotermas de inchamento para os géis de PMDA, com diferentes razões estequiométricas, a 40ºC. 75
Figura (a) 4.8.1.4. - %Seq para os géis de PMDA a diferentes temperaturas. 76
Figura (b)4.8.1.1. - Isotermas de inchamento para os géis de BTDA, com diferentes razões estequiométricas, a 20ºC. 78
Figura (b)4.8.1.2. - Isotermas de inchamento para os géis de BTDA, com diferentes razões estequiométricas, a 30ºC. 78
Figura (b)4.8.1.3. - Isotermas de inchamento para os géis de BTDA, com diferentes razões estequiométricas, a 40ºC. 79
Figura (b)4.8.1.4. - %Seq para os géis de PMDA a diferentes temperaturas. 79
Figura (a) 4.8.2.1.2. - ln(Mt / Meq) como função de ln(T) para o gel 0,25P a diferentes temperaturas. 83
Figura (a) 4.8.2.1.4. - ln(Mt / Meq) como função de ln(T) para o gel 0,75P a diferentes
temperaturas. 83
Figura (a)4.8.2.1.5. - ln(Mt / Meq) como função de ln(T) para o gel 1,0P a diferentes
temperaturas. 84
Figura (b)4.8.2.1.1. - ln(Mt / Meq) como função de ln(T) para o gel 0,5B a diferentes
temperaturas. 85
Figura (b)4.8.2.1.2. - ln(Mt / Meq) como função de ln(T) para o gel 0,75B a diferentes
temperaturas. 86
Figura (b)4.8.2.1.3. - ln(Mt / Meq) como função de ln(T) para o gel 1,0B a diferentes
temperaturas. 86
Figura (a)4.8.2.2.1. - Curva ln(1- Mt / Meq) em função de t para o gel 0,1P a diferentes
temperaturas. 90
Figura (a)4.8.2.2.2. - Curva ln(1- Mt / Meq) em função de t para o gel 0,25P a
diferentes temperaturas. 90
Figura (a)4.8.2.2.3. - Curva ln(1- Mt / Meq) em função de t para o gel 0,5P a diferentes
temperaturas . 91
Figura (a)4.8.2.2.4. - Curva ln(1- Mt / Meq) em função de t para o gel 0,75P a
diferentes temperaturas. 91
Figura (a)4.8.2.2.5. - Curva ln(1- Mt / Meq) em função de t para o gel 1,0P a diferentes
temperaturas. 92
Figura (b)4.8.2.2.1. - Curva ln(1- Mt / Meq) em função de t para o gel 0,5B a
diferentes temperaturas. 94
Figura (b)4.8.2.2.2. - Curva ln(1- Mt / Meq) em função de t para o gel 0,75B a
diferentes temperaturas. 94
Figura (b)4.8.2.2.3. - Curva ln(1- Mt / Meq) em função de t para o gel 1,0B a
diferentes temperaturas. 95
Figura (a)4.8.2.3.1. - Mt/Meq em função da raiz quadrado do tempo, a diferentes
temperaturas para o gel 0,1P. 97
Figura (a)4.8.2.3.2. - Mt/Meq em função da raiz quadrado do tempo, a diferentes
temperaturas para o gel 0,25P. 97
Figura (a)4.8.2.3.3. - Mt/Meq em função da raiz quadrado do tempo, a diferentes
Figura (a)4.8.2.3.4. - Mt/Meq em função da raiz quadrado do tempo, a diferentes
temperaturas para o gel 0,75P. 98
Figura (a)4.8.2.3.5. - Mt/Meq em função da raiz quadrado do tempo, a diferentes
temperaturas para o gel 1,0P. 99
Figura (a)4.8.2.3.6. - Fração linear da curva (Mt/Meq) em função de t 1/2, para o gel
0,1P a diferentes temperaturas. 101
Figura (a)4.8.2.3.7. - Fração linear da curva (Mt/Meq) em função de t 1/2, para o gel
0,25P a diferentes temperaturas. 101
Figura (a)4.8.2.3.8. - Fração linear da curva (Mt/Meq) em função de t 1/2, para o gel
0,5P a diferentes temperaturas. 102
Figura (a)4.8.2.3.9. - Fração linear da curva (Mt/Meq) em função de t 1/2, para o gel
0,75P a diferentes temperaturas. 102
Figura (a)4.8.2.3.10. - Fração linear da curva (Mt/Meq) em função de t 1/2, para o gel
1,0P a diferentes temperaturas. 102
Figura (b)4.8.2.3.1. - Mt/Meq em função da raiz quadrado do tempo, a diferentes
temperaturas para o gel 0,5B. 104
Figura (b)4.8.2.3.2. - Mt/Meq em função da raiz quadrado do tempo, a diferentes
temperaturas para o gel 0,75B. 105
Figura (b)4.8.2.3.3. - Mt/Meq em função da raiz quadrado do tempo, a diferentes
temperaturas para o gel 1,0B. 105
Figura (b)4.8.2.3.4. - Fração linear da curva (Mt/Meq) em função de t 1/2, para o gel
0,5B a diferentes temperaturas. 106
Figura (b)4.8.2.3.5. - Fração linear da curva (Mt/Meq) em função de t 1/2, para o gel
0,75B a diferentes temperaturas. 106
Figura (b)4.8.2.3.6. - Fração linear da curva (Mt/Meq) em função de t 1/2, para o gel
1,0B a diferentes temperaturas. 107
Figura (b)4.8.3.1. - Gráficos de lnD em função de 1/T, para o gel 0,75B. 109
Figura (b)4.8.3.2. - Gráficos de lnD em função de 1/T, para o gel 1,0B. 110
Figura (a)4.8.4.1. - 1/Mt como função de t para os diferentes géis de PMDA a 20ºC.
112
Figura (a)4.8.4.2. - 1/Mt como função de t para os diferentes géis de PMDA a 30ºC.
113
Figura (a)4.8.4.3. - 1/Mt como função de t para os diferentes géis de PMDA a 40ºC.
Figura (b)4.8.4.1. - 1/Mt como função de t para os diferentes géis de BTDA a 20ºC. 115
Figura (b)4.8.4.2. - 1/Mt como função de t para os diferentes géis de BTDA a 30ºC.
116
Figura (b)4.8.4.3. - 1/Mt como função de t para os diferentes géis de BTDA a 40ºC. 116
Figura (a)4.8.5.1. - lnMeq em função de 1/T para o gel 0,1P. 119
Figura (a)4.8.5.2. - lnMeq em função de 1/T para o gel 0,25P. 120
Figura (a)4.8.5.3. - lnMeq em função de 1/T para o gel 0,5P. 120
Figura (a)4.8.5.4. - lnMeq em função de 1/T para o gel 0,75P. 121
Figura (a)4.8.5.5. - lnMeq em função de 1/T para o gel 1,0P. 121
Figura (b)4.8.5.1. - Gráfico de lnMeq em função de 1/T para o gel 0,75B. 122
Figura 4.9.1. - Mecanismo para a síntese do gel de BTDA. 126
Lista de tabelas
Tabela 2.1. - Expoente n da lei de potencia para sistemas de diferentes geometrias. 31
Tabela 4.1.2.1. - Grau de substituição (DS) do Acetol Flakes®. 53
Tabela 4.1.2.2. - Nomes que representarão os géis de BTDA e PMDA. 53
Tabela 4.3.1. - Temperatura de degradação dos diferentes géis. 66
Tabela 4.5.1. - Porosidade, densidade e superfície específica do gel 0,5P e do gel 0,5B.
68
Tabela (a) 4.8.1.1. - Coeficientes angulares e coeficientes de correlação para do gráfico
(%Seq × T) para os géis de PMDA a 20ºC, 30ºC e 40ºC. 76
Tabela (a) 4.8.1.2. - Porcentagem de água no equilíbrio para os géis de PMDA. 76
Tabela(b) 4.8.1.1. - Porcentagem de água no equilíbrio para os géis de BTDA. 79
Tabela (b) 4.8.1.2. - Coeficientes angulares e coeficientes de correlação para o gráfico
(%Seq × T) para os géis de BTDA a 20, 30 e 40ºC. 80
Tabela (a) 4.8.2.1.1. - Valores dos coef. de inchamento para os diferentes géis a
diferentes temperaturas. 84
Tabela 4.8.2.1.1. - Valores dos coef. de inchamento para os diferentes géis a diferentes
temperaturas. 87
Tabela (a)4.8.2.2.1. - Valores da constante da velocidade de relaxação e do coeficiente
de correlação dos géis de PMDA a diferentes temperaturas. 92
Tabela (b)4.8.2.2.1. - Valores da constante da velocidade de relaxação e do coeficiente
de correlação dos géis de BTDA a diferentes temperaturas. 93
Tabela (a)4.8.2.3.1. - Coeficientes de difusão dos géis de PMDA a diferentes
temperaturas. 103
Tabela (b)4.8.2.3.1. - Coeficientes de difusão dos géis de BTDA a diferentes
temperaturas. 107
Tabela (a)4.8.3.1. - Energia de ativação para o processo de difusão para os géis de
Tabela (b)4.8.3.1. - Energia de ativação para o processo de difusão para os géis de
BTDA. 110
Tabela (a)4.8.4.1. - Valores de M`eq, velocidade inicial de inchamento e dos
coeficientes de correlação para as isotermas dos géis de PMDA. 114
Tabela (a)4.8.4.2. - Valores experimentais da quantidade de água absorvida no
equilíbrio (Meq) a diferentes temperaturas e razão entre o valor teórico (M`eq) e o valor
experimental (Meq) da quantidade de água absorvida no equilíbrio. 114
Tabela (b)4.8.4.1. - Valores de M`eq, velocidade inicial de inchamento e dos
coeficientes de correlação para as isotermas dos géis de BTDA. 117
Tabela (b)4.8.4.2. - Valores experimentais da massa de água absorvida no equilíbrio e
razão entre os valores experimentais e os valores teóricos da massa de água absorvida
no equilíbrio. 117
Tabela (a)4.8.5.1. - Valores para os coeficientes de difusão dos géis de PMDA e para
os coeficientes de correlação das retas lnMeq em função de 1/T. 122
Tabela (b)4.8.5.1. - Valores para os coeficientes de difusão dos géis de BTDA e para os
coeficientes de correlação das retas lnMeq em função de 1/T. 123
Capítulo 1
1. Introdução
1.1. A utilização da celulose como matéria prima
Atualmente, o impacto ambiental causado pela crescente demanda de novas tecnologias está entre as grandes preocupações da humanidade. A necessidade de desenvolver materiais biodegradáveis vem sendo objetivo de vários centros de pesquisa e cada vez mais, grandes empresas, seja por imposição governamental ou por estratégia de marketing, despertam interesses em reduzir o impacto ambiental gerado por suas linhas de produção.
Um dos materiais que vem causando grandes impactos ambientais são os polímeros sintéticos. Esses materiais foram produzidos indiscriminadamente nos dois últimos séculos e como conseqüência, a poluição ambiental causada por eles vêm assumindo proporções perigosas. Por um outro lado, materiais poliméricos apresentam propriedades tão peculiares que seria difícil imaginar a nossa vida sem eles.
Na tentativa de reduzir os impactos ambientais gerados por essas macromoléculas e desenvolver novas tecnologias onde suas propriedades são
necessárias, grupos de pesquisa em todo o mundo voltam suas atenções para polímeros naturais, biodegradáveis e renováveis. De um modo geral, um maior número de
pesquisas são destinadas à celulose, ao amido, à quitina e à quitosanaque são polímeros produzidos pela própria natureza e não necessitam do emprego de metodologia
industriais complexas e tão pouco solventes específicos para suas sínteses.
Entre esses polímeros naturais, destacamos aqui a celulose que além de ser biodegradável e renovável é o material natural mais abundante do mundo e o Brasil, sendo um país tropical, apresenta grandes vantagens climáticas que favorecem a produção desse material.
1.2. A produção de celulose no Brasil
exportações frente aos resultados de 2006. Os produtos de celulose e papel brasileiros são fabricados, exclusivamente, a partir de madeira de florestas plantadas, a exemplo do eucalipto e pínus (Associação Brasileira de Celulose e Papel/2007).
Em 2006, as exportações do setor de celulose e papel no Brasil foram de US$ 4,0 bilhões e as previsões para o ano de 2007 são de US$ 4,3 bilhões, esses dados mostram que o setor encontra-se em crescimento (Associação Brasileira de Celulose e Papel/2007).
1.3. O acetato de celulose como matéria prima
Apesar da grande vantagem de ser sintetizada pela natureza, a celulose não é solúvel em solventes orgânicos convencionais e a introdução de grupos menos polares, tais como grupos acetatos, em suas cadeias poliméricas, para a obtenção de novos materiais, é bastante comum.
O acetato de celulose é um material polimérico completamente atóxico que é obtido de uma fonte de celulose de grande pureza como polpa de madeira, linter de algodão ou pasta de celulose, através da reação com ácido acético e anidrido acético utilizando ácido sulfúrico como catalisador, esse processo de acetilação pode ser completo ou parcial. Na acetilação completa a celulose incorpora três grupos de radical acetato por cada unidade de glicose, ao mesmo tempo em que o ácido sulfúrico provoca uma acentuada degradação do polímero de modo que, ao final, apenas 200 – 300
unidades de (β) D-glucopiranose estarão presentes na cadeia do polímero. Alcançada
esta etapa do processo, o acetato de celulose é, em geral, parcialmente hidrolisado pela adição de água até que de 2 – 2,5 grupos de acetato por unidade de glicose permaneçam no polímero. Esse produto recebe o nome comercial de “acetol flakes” (fig. 1.1) e é um polímero termoplástico bastante solúvel em acetona (27% em massa).
O acetato de celulose termoplástico é notável pela sua dureza, maciez ao toque, resistência a impactos, baixa flamabilidade e facilidade de processamento. É usado para fabricação de cabos de chaves de fenda, talheres e escovas, armações de óculos,
lantejoulas, brinquedos, pentes de cabelo e, por sua total atoxicidade, em objetos de contato com o corpo humano.
Acetato de celulose para filtros de cigarro chamado de "filter tow" ou cabo acetato é feito a partir de mantas de acetato de celulose que, por sua vez, são produzidas a partir do "acetol flakes". Este é dissolvido em acetona e regenerado em mantas a partir da evaporação do solvente, a essa manta adiciona-se um plastificante, tri acetato de glicerina, para tornar o produto mais macio, e em seguida puxa-se a mesma por uma máquina de torção. Com a torção a manta se transforma num cilindro poroso que é o filtro de cigarro.
1.4. A produção de acetato de celulose no Brasil
O acetato de celulose é um polímero derivado da polpa de celulose com alto grau de pureza. Em escala comercial, a produção de acetato de celulose é um processo complexo onde a primeira etapa envolve a acetilação da polpa de celulose fazendo uso do anidrido acético para a produção de um produto intermediário conhecido como “floco”. O cabo de acetato é produzido por meio da dissolução dos flocos em acetona. Após a dissolução, o sistema passa por uma filtração para a remoção de partículas e a fiação dos filamentos, que são reunidos para formar uma fita ou corda. Ainda antes de sua comercialização, essa fita passa por um processo no qual é ondulada e condicionada para a remoção dos resíduos de acetona.
1.5. Hidrogéis de acetato de celulose
O uso de uma fonte renovável e biodegradável para a síntese de hidrogéis foi fator predominante na escolha de celulose como matéria prima. A utilização do acetato de celulose ao invés de celulose microcristalina se deve à grande insolubilidade da celulose em solventes orgânicos convencionais, o que torna difícil a síntese dos hidrogéis. É importante lembrar também que o acetato de celulose é um polímero atóxico e essa propriedade possibilita que propostas de bioaplicações sejam feitas para os hidrogéis.
Nesse trabalho, as hidroxilas livres nas cadeias poliméricas do acetato de
celulose foram esterificadas utilizando a trietilamina como catalisador e dois dianidridos como agentes reticuladores, o dianidrido do ácido 1,2,4,5 benzenotetracarboxílico (PMDA) e o dianidrido da 3,3´,4,4´ benzofenonatetracarboxílico (BTDA). As figuras 1.2 e 1.3 mostram a estrutura química dos produtos obtidos após a reação de
esterificação e as figuras 1.4 e 1.5 mostram fotos dos hidrogéis hidratados.
O H O H H H
H O O
H H H H O H O O O O O * * n OH O O O H H H H H O O H H H H OH H O O O O O O * * n O O O Ac Ac Ac Ac Ac Ac Ac Ac Ac Ac
O OH O
O O OH O
O H O
H H
H
H O O
H H
H H O H
O O
O
O O
* *
O H O
H
H H
H O O
H
H H
H O H
O O
O
O O
* *
Ac
Ac Ac
Ac Ac
Ac
Ac
Ac Ac
Ac
Figura 1.3. Estrutura química do hidrogel de BTDA.
Figura 1.4. Hidrogéis de acetato de celulose reticulados com PMDA. Na figura A mostramos o gel 0,1P com ampliação de 8X, na figura B mostramos o gel 1,0P com uma ampliação de10X, na figura C mostramos o gel 0,25P com ampliação de 50X e na figura D mostramos o gel 0,25P com ampliação de 6,5X.
Figura 1.5. Hidrogéis de acetato de celulose reticulados com BTDA. Na figura A mostramos o gel 0,5B com ampliação de 6,5X, na figura B mostramos o gel 0,5B com ampliação de 8X e na figura C
Como pode ser visto nas figuras 1.4 e 1.5 a reação de esterificação transforma as cadeias poliméricas do acetato de celulose em redes poliméricas hidrofílicas.
Nesse trabalho, descrevemos o início de um estudo que tem como objetivo caracterizar e propor aplicabilidades para esses hidrogéis de acetato de celulose
1.6. Objetivos
1.6.1. Gerais
Este trabalho teve como objetivo geral, sintetizar e caracterizar géis de acetato de celulose, obtidos através da reticulação das cadeias poliméricas com anidrido
piromelítico (PMDA) e o anidrido do ácido 3,3´,4,4´ benzofenona tetracarboxílico (BTDA), visando obter um material capaz de absorver grandes quantidades de água.
1.6.2. Específicos
• Caracterizar as matérias primas e os produtos por técnicas de
espectroscopia na região do infravermelho (FTIR), termogravimetria (TGA), calorimetria exploratória diferencial (DSC) e microscopia eletrônica de varredura (MEV).
• Analisar o grau de reticulação das amostras por espectroscopia no
ultravioleta (UV)
• Determinar a microporosidade dos géis e a densidade empregando a
técnica de BET.
• Obter isotermas de absorção de água a 20, 30 e 40ºC.
• Determinar o mecanismo de absorção de água pelos hidrogéis.
• Determinar a cinética de inchamento para os hidrogéis.
• Determinar a entalpia de mistura do sistema hidrogel/água.
Capítulo 2
2. Revisão bibliográfica
2.1. Celulose
A celulose é um homopolímero natural (poli-β(1,4)-D-glucose), não ramificado,
fibroso, com configuração sindiotática e insolúvel em água. Esse é o material natural mais abundante do mundo e o principal componente da madeira e das fibras vegetais. Ele é composto de unidades de (β) D-glucopiranose (fig. 2.1.1), unidas através de ligações glicosídicas entre os carbonos 1 e 4. Na cadeia de celulose cada mero de (β) D-glucopiranose se encontra girado de 180º em relação à próxima unidade. (OSullivan, 1997) (Hon, 1994) (Albert L.Lehninger et al., 1995) (R.Boyd and R.Morrison, 1983)
O
C1
C4
C5
H
HO
H HO
H
H OH H
C6
OH C2
C3
OH
Figura 2.1.1. Unidade de (β) D-glucopiranose
Para uma melhor caracterização da celulose é bastante útil examina-la em
diferentes níveis estruturais. O nível estrutural primário, que diz respeito ao arranjo dos átomos na unidade monomérica de glucopiranose e consiste na estrutura apresentada na figura 2.1.1. O nível estrutural secundário que consiste no arranjo das unidades
monoméricas dentro da macromolécula e o nível estrutural terciário, que diz respeito aos arranjos estruturais das moléculas de celulose uma em relação à outra (Atalla R.H., 1999).
O arranjo estrutural primário encontra-se elucidado há algum tempo e não existem grandes questões a seu respeito. Quanto aos arranjos estruturais secundário e terciário, apesar da grande quantidade de estudos feitos para tentar elucidá-los, um arranjo definitivo encontra-se ainda em discussão (Atalla R.H, 1999).
Estudos baseados em espectroscopia Ramam e RMN 13C do estado sólidorevelam
unidades de (β) D-glucopiranose não são simetricamente equivalentes e, por isso, a unidade de repetição básica da celulose, deveria ser considerada a anidrocelobiose (fig. 2.1.2). Um suporte para essa idéia é apresentado por Kono, H/2004 em seus estudos de elucidação da estrutura cristalina da celulose II. Nesse trabalho, o autor evidencia a existência de dois diferentes tipos de resíduos de anidroglucose (Atalla R.H., 1999) (Kono and Numata, 2004).
O H
O
H HO
H
H OH H HO
O H
O H HO
H H OH H
HO
O H
O H HO
H H OH H
HO *
* n
Figura 2.1.2. A estrutura circulada pela linha pontilhada representa a unidade de anidrocelobiose.
Em seu nível estrutural terciário, a celulose se revela um polímero altamente cristalino. Estimativas das proporções de cristalinidade dão valores entre 50% e 90%. Alguns autores atribuem valores de até 95% de cristalinidade. É importante ressaltar que a porcentagem de cristalinidade em uma determinada amostra de celulose varia com a técnica empregada para medida da cristalinidade, origem da amostra, processo de purificação, tratamento recebido pela amostra, etc (Hon, 1994).
2.2. Estruturas cristalinas da celulose
A celulose possui diferentes formas cristalinas (polimorfismo), que foram
constatadas por ressonância magnética nuclear (RMN), espectroscopia do infravermelho e estudos de difração. Esses estudos mostraram que ela possui seis estruturas
polimórficas, que recebem os seguintes nomes: celulose I, celulose II, celulose III I,
celulose IIIII, celulose IVI e celulose IVII (OSullivan, 1997) (Hon, 1994).
Das seis formas polimórficas, a mais estudada é a celulose I, que é a celulose nativa, isto é, na forma como ela é encontrada na natureza. A celulose II, a segunda forma polimórfica mais estudada, pode ser obtida através de dois processos: 1) mercerização, que consiste em um tratamento com NaOH ou 2) regeneração, que consiste em solubilizar a celulose em algum solvente, e em seguida, precipita-la
pelo tratamento, da celulose I e II, com amônia ou amina. Obtemos as celuloses IV I e
IVII aquecendo, respectivamente, as celuloses III I e III II, em glicerol, a 206 ºC
(OSullivan, 1997) (Lindgren et al., 1995) (Ishikawa et al., 1997).
Usando CP/MAS, 13CRMN, FTIR e difração de elétrons (DE), constatou-se a
existência de duas formas polimórficas para a celulose I que são chamadas de: celulose Iα e Iβ. A quantidade de cada uma dessas fases, α e β, em uma determinada amostra, varia de acordo com a origem do material (Hult et al., 2003) (OSullivan, 1997) (Lindgren et al., 1995) (Hon, 1994) (Hardy and Sarko, 1996).
Duas linhas de investigações são utilizadas para explicar as diferenças entre as duas formas alomorfas da celulose I. Uma delas se baseia em estudos difratométricos, que indicam que a forma Iα possui estrutura triclínica, com uma cadeia por célula unitária, e sem unidades equivalentes de anidroglucose. Os estudos sugerem que a forma Iβ possui células monoclínicas de grupo espacial P2,com duas cadeias por célula unitária e eixos em dupla hélice, simetricamente coincidentes com as cadeias, obrigando que as unidades adjacentes de anidroglucose sejam simetricamente equivalentes. A outra linha de estudos se baseia em estudos espectrométricos, que apontam para similares estruturas secundárias, que se diferenciam apenas nas diferentes ligações de hidrogênio entre as cadeias (Atalla R.H., 1999).
A fase Iα é termodinamicamente mais estável e pode ser transformada, quase
inteiramente, em Iβ, por tratamento térmico. Apesar da grande quantidade de estudos com as diferentes formas alomorfas da celulose I, as diferenças entre elas não estão ainda completamente esclarecidas (Lindgren et al., 1995) (Atalla R.H., 1999).
Durante a transformação polimórfica da celulose I para celulose II, através da mercerização ou regeneração, o empacotamento cristalino da forma nativa, cadeias paralelas, se rearranja em um empacotamento antiparalelo, característico da forma polimórfica II. Essa transformação polimórfica é irreversível e normalmente
acompanhada por uma diminuição na cristalinidade do material. Processos, in vitro, indicam que a forma termodinamicamente favorável é a forma polimórfica da celulose II (Ass et al., 2006) (OSullivan, 1997).
A estrutura da celulose II possui duas cadeias por unidade de célula e um grupo
espacial P21. Estudos de espalhamento de nêutrons, RMN 2D spin-exchange e dados de
difração síncrotrons da celulose II confirmam o arranjo antiparalelo proposto por
esqueleto, mas similaridades nas conformações dos grupos hidroximetil (Kono and Numata, 2004).
A classificação, cadeias paralelas e antiparalelas, utilizada anteriormente, referem às seguintes definições: quando dentro de uma mesma célula unitária, as cadeias de
celulose apresentam a mesma direção das ligações glicosídicas 1 4 diz-se que as
cadeias são paralelas. Se uma das cadeias possui uma ligação glicosídica,
correspondente à direção positiva do eixo da fibra, e a segunda cadeia têm a ligação no sentido negativo do eixo da fibra, o arranjo dessas cadeias é dito antiparalelo
(OSullivan, 1997).
As celuloses IIII e IIIII são obtidas, reversivelmente, a partir das celuloses I e II,
respectivamente, enquanto as formas alotrópicas IV I e IV II são obtidas,
irreversivelmente, a partir das celuloses IIII e IIIII, respectivamente (OSullivan, 1997).
A estrutura altamente cristalina e organizada da celulose se deve aos grupos hidroxilas, que ficam a mostra nas cadeias poliméricas lineares e formam ligações de hidrogênio intra- e intermolecular, como pode ser observado na figura 2.2.1. Devido à essa alta cristalinidade natural, a celulose não é solúvel em solventes convencionais e a solubilidade das fibras de celulose vem apenas de seus segmentos amorfos e/ou
componentes não celulosídicos (Boluk, 2005) (Ass et al., 2006).
O H H OH H H HO H O OH O H H OH H H HO H HO O H O H OH H H HO H O OH O H H OH H H HO H O OH O H H OH H H HO H OH O H O H OH H H HO H O OH O H H OH H H HO H O OH O H H OH H H HO H HO O H O H OH H H HO H O OH O H H OH H H HO H O OH O H H OH H H HO H OH O H O H OH H H HO H O OH n n
Figura 2.2.1. Algumas das possíveis ligações de hidrogênio intra e intermolecular formadas entre as cadeias de celulose.
2.3. As regiões amorfas da celulose
formam as macrofibrilas. As micro e macrofibrilas são as unidades de construção das fibras de celulose (OSullivan, 1997) (Boluk, 2005).
Existem poucas informações estruturais das regiões amorfas da celulose, quando essas são comparadas com o grande número de informações vindas da caracterização de sua estrutura cristalina. Isto talvez se deva à terminologia, que sugere que, as regiões amorfas são completamente sem estrutura, e também devido aos métodos de
caracterização serem limitados à WAXD e CP-MAS 13C NMR, para medir a presença
de fase amorfa. Alguns estudos da fase amorfa da celulose indicam a presença de certos “arranjos” estruturais, formados, devido às ligações de hidrogênio intra- e
intermolecular (Kondo and Sawatari, 1996) (Hishikawa et al., 1999).
Como a interação solvente/celulose se dá em regiões amorfas da cadeia, uma vez que as regiões altamente empacotadas são muito densas e inacessíveis aos solventes, a caracterização da fase amorfa é de extrema importância, pois, ela pode ser o primeiro passo na descoberta dos detalhes do arranjo das moléculas quando em solução (Kondo and Sawatari, 1996) (Morgenstern and Kammer, 1999) (Boluk, 2005) (Ass et al., 2006).
Visando melhorar o acesso do solvente às cadeias de celulose, é comum o
emprego de técnicas, onde as cadeias são desintegradas em uma determinada extensão, usando ar ionizado (Ass et al., 2006), tratamentos termo mecânicos, ativação
enzimaticamente controlada, etc (Morgenstern and Kammer, 1999).
Segundo Ass e Frollini, a ordem de reatividade dos grupos hidroxilas na cadeia de celulose é C6 >>C2>C3. Essa ordem de reatividade foi observada também, quando as cadeias de celulose estão em solução. Confirmando que, mesmo quando a celulose aparece visualmente como um sistema homogêneo, pode existir algum grau de
agregação e/ou associação das cadeias poliméricas dissolvidas a um nível coloidal (Ass et al., 2006).
2.4. Ésteres de Celulose
As reações da celulose em que a cadeia polimérica fica intacta se dão nos grupos hidroxilas livres presentes nas unidades de glucose. Estas reações utilizadas para modificar as propriedades de um polímero, já pronto, barato e facilmente acessível possuem excepcional importância industrial (R.Boyd and R.Morrison, 1983).
Os primeiros derivados de celulose sintetizados em laboratório e produzidos em escala industrial foram os ésteres de celulose de ácidos orgânicos e inorgânicos. Esses ésteres se formam quando os grupos hidroxilas das cadeias de celulose são substituídos por grupos acila. A diminuição da quantidade de grupos –OH na cadeia de celulose, leva a formação de derivados, normalmente mais solúveis em solventes convencionais, um vez que, passamos a trabalhar com materiais menos cristalinos devido a diminuição das possibilidades de ligação de hidrogênio intra e intermolecular (Freire et al., 2005).
Os ésteres de celulose mais importantes produzidos industrialmente são acetato de celulose (CA), propionato acetato de celulose (CAP) e butirato acetato de celulose (CAB). Esses ésteres de celulose, mesmo sendo, normalmente, mais caros que termoplásticos originados de petróleo, são produzidos em grande escala por causa de suas excelentes propriedades (Adair Rangel de Oliveira Júnior, 2002).
Tanto a natureza do grupo substituinte, tanto a extensão em que os grupos hidroxilas são substituídos por esses grupos, levam a diferentes propriedade térmicas, mecânicas e físico químicas. Para saber a extensão da substituição das hidroxilas por grupos acilas em um determinado éster de celulose, usa-se o termo grau de substituição (DS), que corresponde ao número médio de grupos substituintes por unidade de
anidroglucose. Os valores do grau de substituição podem varia de valores próximos a 0 até o valor máximo de substituição 3 (Adair Rangel de Oliveira Júnior, 2002; Freire et al., 2005; Samios et al., 1997).
A potencial biodegradabilidade de ésteres de celulose é investigada por vários autores e ainda se encontra em discussão. Segundo E. Samios et al. /1997, a
biodegradabilidade do acetato de celulose depende do grau de substituição do derivado celulósico. No trabalho desenvolvido por D. Krishna Bhat, foi mostrado que a natureza dos grupos substituintes na cadeia de celulose afeta a biodegradabilidade de blendas obtidas com diferentes razões de esteres de celulose (acetato de celulose e ftalato acetato de celulose) e PMMA. Um exame cuidadoso da literatura corrente mostra que muitos dos ésteres de celulose apresentam algum grau de biodegradabilidade e que essa biodegradabilidade é uma grande vantagem frente a materiais poliméricos não
biodegradáveis, uma vez que, o impacto ambiental causado por materiais poliméricos utilizados em nosso dia-a-dia é objeto de crescente avaliação (Adair Rangel de Oliveira Júnior, 2002; D.Krishna Bhat and M.Selva Kumar, 2006; Samios et al., 1997).
O estudo de polímeros biodegradáveis é um campo emergente, pois, a disposição dos mesmos no meio ambiente não causa qualquer risco à população (D.Krishna Bhat and M.Selva Kumar, 2006).
Ésteres de celulose vêm sendo empregados na fabricação de fibras têxteis, materiais plásticos, filmes, blendas poliméricas, excipientes e cápsulas para a indústria farmacêutica, filtros de cigarro, reforço em materiais compósitos, etc (Adair Rangel de Oliveira Júnior, 2002; D.Krishna Bhat and M.Selva Kumar, 2006; Freire et al., 2005; Jessica D.Posey-Dowty et al., 2007; Kevin J.Edgar, 2007; Saake et al., 2006).
2.5. Acetato de Celulose
O acetato de celulose (AC) é um polímero de grande importância industrial, sua utilidade inclui fios de algodão para a indústria têxtil, filtros, filmes fotográficos
transparentes e pigmentados e na composição de plásticos, como aqueles utilizados para compressão, extrusão, injeção e, em menor extensão, para revestimento de superfícies. Esse é um polímero amorfo, não tóxico e inodoro (fig. 2.5.1). É resistente a ácidos fracos, estável em óleos minerais e permeável a vapor d`água e a álcool, sendo um material termoplástico que possui custo reduzido (Samios et al., 1997).
foi um dos poucos polímeros utilizados na separação e purificação de gases (Adair Rangel de Oliveira Júnior, 2002).
A produção desse importante derivado de celulose, no Brasil, é feita pela empresa Rhodia do grupo francês Rhone Poulenc, existindo ainda outros 10 fabricantes no mundo (Adair Rangel de Oliveira Júnior, 2002; Jessica D.Posey-Dowty et al., 2007; Kevin J.Edgar, 2007).
Um fator muito importante na biodegradabilidade do acetato de celulose é o seu grau de substituição, a biodegradação se torna mais fácil para o acetato menos
substituído e um aumento no grau de substituição leva a uma diminuição na
cristalinidade do material. O grupo acetil ligado ao carbono 6 é o mais vulnerável à hidrólise, seguido pelo grupo ligado ao carbono 2 e finalmente o grupo ligado ao carbono 3 (Samios et al., 1997; Sh.S.Arslanov et al., 1999).
Se considerando a afinidade dos grupos que constituem a cadeia polimérica do acetato de celulose com a água, conclui-se que os grupos acetil são hidrofóbicos e os grupos hidroxila são hidrofílicos e podem formar ligações de hidrogênio com a água. Acetato de celulose com grau de substituição 2,5, apresenta aproximadamente 16% em massa de água absorvida, quando este se encontra em equilíbrio com a água
(K.C.Khulbe et al., 2004).
2.6. Redes poliméricas
Redes poliméricas são redes moleculares, formadas por ligações covalentes e ou interações físicas entre macromoléculas. Essas interações entre as macromoléculas recebem o nome de ponto de junção. É importante ressaltar que, em redes poliméricas, pontos adjacentes de junção são separados por subcadeias normalmente lineares, formadas por muitas ligações covalentes.
As propriedades de uma rede polimérica são determinadas pela estrutura e pelo comprimento das sub-redes entre os pontos de junção, pela estrutura e comprimento das cadeias que formam os pontos de junção e pelo tipo de interação entre as
macromoléculas. É essencial para a formação de redes poliméricas a presença de pelo menos um reagente com funcionalidade maior do que dois (f >2). Essa propriedade leva à ramificação da estrutura molecular e formação da rede polimérica (R.F.T.Stepto, 1998).
Em uma reação de polimerização, o número de grupos reativos de uma
macromolécula cresce com o aumento do número de ramificações, levando à formação de redes complexas. A ramificação da estrutura pode ser precisamente controlada e o crescimento da rede é limitado apenas pelo volume do meio reacional (R.F.T.Stepto, 1998).
Existem dois processos que levam a formação da rede polimérica, o
entrecruzamento ou reticulação (crosslinking) e a polimerização de grupos terminais (endlinking).
A formação da rede através da polimerização de grupos terminais se dá quando grupos reativos estão no final da cadeia ou subcadeia. Veja figura 2.6.1.
Figura 2.6.1. Formação de uma rede polimérica através da polimerização de grupos terminais.
Figura 2.6.2. Formação de uma rede polimérica através da reticulação das cadeias.
A presença de elevados valores de funcionalidade nos reagentes leva a gelificação e freqüentemente a grandes quantidades de ciclização (reação intramolecular).
Em uma reação de polimerização não linear, o ponto onde a continuidade através do volume reagente primeiro aparece, é denominado ponto de gel. Flory e Stockmayer definiram, quantitativamente, em termos de conversão química, o ponto de gel como o ponto na reação de polimerização onde a continuidade molecular através do volume reagente ocorre com probabilidade 1. Normalmente o crescimento da rede polimérica vai além do ponto de gel (R.F.T.Stepto, 1998).
2.7. Hidrogéis
De uma maneira geral, os processos de entrecruzamento e grafitização são
utilizados para melhorar as propriedades de polissacarídeos, formando redes poliméricas tridimensionais que incham rapidamente, absorvendo uma grande quantidade de água, segundo Mostafa & Morsy/2004, citado por Baljit Singh et al./2006. Esses
processamentos acabam por transformar cadeias poliméricas lineares em géis ou hidrogéis (Baljit Singh et al., 2006).
Yoshimura et al., 2005). Alguns géis podem absorver proporções extremamente altas de água e são chamados de géis “superabsorventes” (Erdener Karadag et al., 2006).
Propostas para a aplicabilidade desses materiais dependem principalmente da sua capacidade de absorver água, essa capacidade de inchamento vem da presença de
grupos hidrofílicos como –OH, -CONH-, -COOH, -SO 3H e –CONH 2 em sua rede
polimérica. De acordo com a natureza dos grupos que formam a rede polimérica, os hidrogéis podem ser classificados como iônicos ou neutros e baseando na estrutura física da rede, hidrogéis podem ser classificados como amorfos, semicristalinos ou reticulados por ligações de hidrogênio, por estruturas supramoleculares ou agregados hidrocoloidais (Nikolaos A.Peppas and Atul R.Khare, 1993).
Hidrogéis podem ser sintetizados pelos mesmos métodos utilizados para a preparação de materiais poliméricos em geral e utilizando as mesmas técnicas. As reações de reticulação são feitas introduzindo um ou mais monômeros multifuncionais (agente reticulante) durante o crescimento das cadeias poliméricas ou entre duas ou mais cadeias poliméricas já sintetizadas anteriormente. A reação de polimerização pode ser iniciada utilizando radicais livres gerados por aquecimento, ionização, radiação e/ou por agentes redutores ou oxidantes (A.El-hag Ali et al., 2007; Baljit Singh et al., 2006; Carlos Peniche et al., 1997; Erdener Karadag et al., 2002; Erdener Karadag et al., 2004b; Erdener Karadag et al., 2004a; Esmaiel Jabbari and Samyra Nozari, 2000; Ko-Shao Chen et al., 2005; Laura Serra et al., 2007; Nikolaos A.Peppas and Atul R.Khare, 1993; S.Benamer et al., 2006; S.K.Bajpai and Susamma Johnson, 2005).
Durante a síntese de hidrogéis a presença de solvente no meio reacional, bem como o tipo de solvente no meio, influenciam diretamente nas propriedades do gel. Se a quantidade de água presente durante a polimerização é maior do que a capacidade de absorção do gel no equilíbrio, uma separação de fase durante a formação da rede ocorre e uma rede polimérica heterogênea é formada, consistindo de domínios altamente reticulados (microgéis) conectados por cadeias pouco reticuladas. Esse fenômeno é conhecido como microsinerese. A quantidade de solvente no meio reacional é tão importante que um hidrogel sem poros pode se tornar poroso variando apenas essa condição reacional (Nikolaos A.Peppas and Atul R.Khare, 1993).
origem aos hidrogéis físico. Hidrogéis formados através de ligações covalentes, ou seja, a reticulação das cadeias poliméricas é feita através de ligações interatômicas, são chamados de hidrogéis químicos (Nikolaos A.Peppas and Atul R.Khare, 1993; S.Benamer et al., 2006).
Os processos aos quais um hidrogel foi submetido durante sua síntese ou processamento têm grande influência no seu comportamento de inchamento. Após ter sido inchado até o equilíbrio e posteriormente seco, o mecanismo, a cinética de inchamento e até mesmo a estrutura do hidrogel podem mudar. Uma explicação para esse fenômeno é feita baseando na separação das cadeias que se encontravam
emboladas antes do processo de inchamento (Nikolaos A.Peppas and Atul R.Khare, 1993).
Em todos os casos, o desempenho do gel em determinada aplicação depende de detalhes em sua estrutura. Freqüentemente, um único valor numérico, como razão de inchamento ou uma densidade efetiva de agente entrecruzante é empregada para
caracterizar a rede estrutural do hidrogel, citado por Dimitri R. Kioussis/2005, segundo Kofinas P./1997. Uma caracterização quantitativa e mais detalhada da rede polimérica entrecruzada permanece um problema, apesar de haver sido feitos avanços na
explicação estrutural de polímeros lineares solúveis (Dimitri R.Kioussis and Peter Kofinas, 2005). A insolubilidade da rede e suas complexidades espaciais e topológicas são as principais razões para a dificuldade na sua caracterização (Dimitri R.Kioussis and Peter Kofinas, 2005).
O grau de reticulação na rede polimérica, que é uma medida da quantidade de cadeias entrecruzantes presentes no material, é estudado sob vários aspectos, entre eles em propriedades como influência na transição de fase a diferentes pH e temperatura (Trong-Ming Don, Hann-Ru Chen/2005) e influência na estrutura tridimensional da rede e no percentual de inchamento (Baljit Singh et al./2006).
Segundo Dimitri R. Kioussis, Peter Kofinas/2005 a quantidade de agente
2.7.1. Hidrogéis neutros
Hidrogéis neutros são hidrogéis que não apresentam grupos ionizáveis na rede polimérica e devido a isso não sofrem influências de variações de pH e força iônica do meio. Normalmente, esses hidrogéis são sensíveis às variações na temperatura do meio reacional. Um aumento na temperatura aumenta a mobilidade das cadeias poliméricas e conseqüentemente alteram a capacidade de inchamento dos mesmos. Esses hidrogéis encontram grande utilidade em sistemas onde não se deseja variação nas propriedades do hidrogel frente à variação na força iônica do meio bem como no pH (Nikolaos A.Peppas and Atul R.Khare, 1993).
A interação da água ou solventes de grande afinidade com a rede polimérica, em hidrogéis neutros, leva a uma tendência das cadeias poliméricas a se solubilizarem aumentando assim a entropia do sistema, essa solubilização das cadeias é impedida pelos pontos de entrecruzamento da rede, que atuam como forças retrativas elásticas e impedem essa dissolução. Estes dois fatores se contrabalanceiam quando um hidrogel neutro interage com um solvente (Nikolaos A.Peppas and Atul R.Khare, 1993).
Flory e Rehner desenvolveram um modelo do inchamento de géis reticulados utilizando uma distribuição Gaussiana das cadeias poliméricas. Eles desenvolveram um modelo para descrever o grau de equilíbrio de polímeros reticulados, postulando que o grau de inchamento de uma rede polimérica é governado pelas forças elásticas retrativas das cadeias polimérica e pela compatibilidade termodinâmica da rede e das moléculas de água. Em termos de energia livre do sistema, a variação total da energia livre no inchamento pode ser descrita como:
∆G = ∆Gelástica + ∆Gmistura (eq. 2.1)
onde ∆Gelástica é a contribuição da força retrativa elástica e ∆Gmistura representa a
compatibilidade termodinâmica do polímero e do solvente.
Mantendo constante a temperatura e a pressão, podemos derivar uma expressão para a mudança no potencial químico da água em termos das contribuições devido ao inchamento.
onde ∝g é o potencial químico do solvente dentro do gel e ∝p é o potencial químico do
solvente puro. Como no equilíbrio o potencial químico do solvente dentro do gel e do solvente puro são iguais, a contribuição elástica e da mistura para o potencial químico irão se contrabalancear.
O fator atribuído ao processo de mistura pode ser calculado através do calor de mistura e da entropia de mistura e pode ser expresso através da seguinte equação:
∆∝mistura = RT(ln(1-v2s) + v2s + χ1v2s ) (eq. 2.3)
onde v2s é a fração volumétrica do polímero e χ1 é o parâmetro de interação solvente
polímero.
A contribuição elástica para o potencial químico é determinada pela teoria estatística da elasticidade da borracha. A energia livre elástica depende do número de cadeias na rede e do fator de expansão linear. Para géis que são reticulados na ausência de água, a contribuição para o potencial químico é escrita como:
∆∝elástico = (V1/vMc)(1-{2Mc/Mn})((v2s )1/3 – v2s/2) (eq. 2.4)
onde v é o volume específico do polímero, V 1 é o volume molar do solvente, M c é a
massa molecular média entre dois pontos de junção e M n é a massa molecular das
cadeias poliméricas sem a presença do agente reticulador. Para polímeros reticulados na presença de água a equação para a contribuição elástica para o potencial químico é:
∆∝elástico = RT(V1/vMc)(1-{2Mc/Mn}) v2r ((v2s/ v2r )1/3 – (v2s/2 v2r)) (eq. 2.5)
onde v 2r é a fração volumétrica do polímero em seu estado relaxado, isto é,
imediatamente após a reticulação. Sendo assim, a contribuição termodinâmica para géis neutros reticulados na ausência e presença de água podem ser descritas,
respectivamente, pelas seguintes equações.
1/Mc = 2/ Mn – {(v/V1)[ln(1- v2s) + v2s + χ1v2s]}/ v2r ((v2s/ v2r )1/3 – (v2s/2 v2r)) (eq. 2.7)
2.7.2. Hidrogéis iônicos
Hidrogéis iônicos são obtidos através da utilização de monômeros iônicos na síntese de polímeros, monômeros iônicos com monômeros iônicos ou não iônicos na síntese de copolímeros ou através da utilização de agentes reticuladores que possuam grupos iônicos (Nikolaos A.Peppas and Atul R.Khare, 1993).
Esses hidrogéis podem ser classificados em aniônicos, catiônicos e anfóteros, de acordo com os grupos presentes em suas cadeias. Géis aniônicos possuem grupos funcionais capazes de apresentarem cargas negativas, tais como ácido carboxílico e ácido sulfônico, a ionização desses géis acorre quando o pH da solução se encontra
acima do pK a dos grupos ionizáveis presentes na rede polimérica. Géis catiônicos
possuem grupos funcionais tais como aminas que se ionizam em meios com pH menor que o pKb das espécies ionizáveis. No caso de hidrogéis anfóteros que são hidrogéis que
possuem grupos básicos e ácidos em sua estrutura, haverá grupos ionizados em pH muito ácido e muito básico, no ponto de pH isoelétrico a capacidade absortiva do hidrogel é moderada (Nikolaos A.Peppas and Atul R.Khare, 1993).
Hidrogéis iônicos apresentam vantagens quando comparados com hidrogéis neutros, pois como são sensíveis ao pH e força iônica da solução, apresentam uma grande variedade de aplicações como materiais biomédicos (Nikolaos A.Peppas and Atul R.Khare, 1993).
O grau de absorção de água por esses hidrogéis sofre forte influência de fatores como a força iônica e o pH da solução. Isso se deve à protonação ou desprotonação de grupos ácidos ou básicos presentes na rede polimérica que acabam por promover o afastamento das cadeias poliméricas e aumentar a capacidade desses materiais em absorver água. Hidrogéis iônicos absorvem, normalmente, maiores quantidades de água do que hidrogéis neutros. Um aumento no conteúdo iônico de uma rede polimérica torna a rede mais hidrofílica e conseqüentemente leva a uma maior velocidade de
absorção e uma maior capacidade de inchamento (Nikolaos A.Peppas and Atul R.Khare, 1993).