Hypercholesterolemia promotes bone marrow cell mobilization by perturbing the
SDF-1:CXCR4 axis
Ana L. Gomes,1-3Taˆnia Carvalho,1-3Jacinta Serpa,1-3Cheila Torre,1-3and Se´rgio Dias1-3
1Angiogenesis Group, Instituto Portugueˆs de Oncologia de Francisco Gentil, Centro de Lisboa, EPE (CIPM/IPOLFG), Lisboa;2Instituto Gulbenkian de Cieˆncia, Oeiras; and3CEDOC, Faculdade de Cieˆncias Me´dicas, Universidade Nova de Lisboa, Lisboa, Portugal
Hypercholesterolemia is associated with elevated peripheral blood leukocytes and increased platelet levels, generally attrib-uted to cholesterol-induced proinflamma-tory cytokines. Bone marrow (BM) cell mobilization and platelet production is achieved by disrupting the SDF-1:CXCR4 axis, namely with granulocyte colony-stimulating factor and/or CXCR4 antago-nists. Here we show that high cholesterol disrupts the BM SDF-1:CXCR4 axis; pro-motes the mobilization of B cells, neutro-phils, and progenitor cells (HPCs); and creates thrombocytosis. Hypercholester-olemia was achieved after a 30-day
high-cholesterol feeding trial, resulting in el-evated low-density lipoprotein (LDL) cholesterol levels and inversion of the LDL to high-density lipoprotein choles-terol ratio. Hypercholescholes-terolemic mice dis-played lymphocytosis, increased neutro-phils, HPCs, and thrombocytosis with a lineage-specific decrease in the BM. His-tologic analysis revealed that megakaryo-cyte numbers remained unaltered but, in high-cholesterol mice, they formed large clusters in contact with BM vessels. In vitro, LDL induced stromal cell–derived factor-1 (SDF-1) production, suggesting that megakaryocyte delocalization
re-sulted from an altered SDF-1 gradient. LDL also stimulated B cells and HPC mi-gration toward SDF-1, which was blocked by scavenger receptor class B type I (cholesterol receptor) inhibition. Accord-ingly, hypercholesterolemic mice had in-creased peripheral blood SDF-1 levels, increased platelets, CXCR4-positive B lymphocytes, neutrophils, and HPCs. High cholesterol interferes with the BM SDF-1:CXCR4 axis, resulting in lymphocy-tosis, thrombocylymphocy-tosis, and HPC mobiliza-tion. (Blood. 2010;115(19):3886-3894)
Introduction
Bone marrow (BM) cells have a well-defined and consistent lineage-specific spatial distribution of hematopoietic progenitor (HPC) and maturing cells, mainly between 2 locations, also termed niches: the endosteal and the vascular niche.1-3The BM endosteal niche, which mainly lodges the stem cell population, is located in the inner surface of the bone.1The BM vascular niche is repre-sented by the BM sinusoidal microvasculature with adjacent megakaryocytes (MKs)4and surrounding hematopoietic cells (HCs) and allows the exit of mature HCs to the peripheral blood (PB), being the site where HPCs differentiate and set the stage for full reconstitution of hematopoiesis after BM ablation.2
External stimuli, which alter hematologic parameters, act most likely by disturbing the homeostasis of the BM niches, thereby interfering with the spatial distribution of HCs. Namely, increased systemic cholesterol levels (hypercholesterolemia) are known to induce endothelial activation by increased expression of adhesion molecules and the release of proinflammatory cytokines, which are believed to result in leukocyte recruitment3,5,6and may explain the leukocytosis observed in patients with hypercholesterolemia.7 However, in patients with hypercholesterolemia, thrombocytosis (high platelet count) is a common finding, and MKs were shown to be altered.7-11However, the mechanistic basis for thrombocytosis in patients with hypercholesterolemia is yet to be elucidated.
We have previously shown that the mobilization of MKs and consequent platelet production (thrombopoiesis) is achieved through
stromal cell–derived factor-1 (SDF-1)–mediated MK migration.12 Therefore, expression of CXCR4 by mature MKs is one of the major signaling pathways involved in the transmigration of MK.13,14 Here, by using a murine model of hypercholesterolemia, we demonstrate that the use of a high-cholesterol diet increased cholesterol systemic levels and was associated with alterations of the BM vascular niche, inducing SDF-1 production and promoting the exit of B lymphocytes and neutrophils from the BM. Hypercho-lesterolemia also is associated with massive MK delocalization to the BM vessels lining, which clinically translates into thrombocyto-sis. Taken together, the data presented in this work suggest that high cholesterol induces BM-cell mobilization and platelet production by interfering with the SDF-1:CXCR4 axis.
Methods
Animals and experimental design
All animal experiments were performed with the approval of the Instituto Gulbenkian de Ciencia Animal Care Committee and Review Board. C57Bl/6j mice (male and female, 4-6 weeks old) were subject to a 30-day high-cholesterol feeding trial. In brief, groups of 6 animals were put on a high-fat/high-cholesterol/cholate diet (15%/1.25%/0.5%, respectively), with food and water ad libitum; animals were killed after 30 days on the diet. In parallel, groups of 6 animals were fed the standard mouse diet and were used as controls.
Submitted August 27, 2009; accepted November 18, 2009. Prepublished online as Blood First Edition paper, December 15, 2009; DOI 10.1182/blood-2009-08-240580.
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked ‘‘advertisement’’ in accordance with 18 USC section 1734.
© 2010 by The American Society of Hematology
Sample collection and morphologic analysis
Before sacrifice, blood was collected from the cheek pouch of the mice, put into in EDTA (ethylenediaminetetraacetic acid)–coated tubes (Multivette 600; Sarstedt), and analyzed with a Hemavet 950FS cell counter (Drew Scientific). BM was removed in toto by flushing 1 femoral cavity, and BM smears were prepared by streaking the exposed BM onto a glass slide. Pressure while executing the smears was adjusted to disperse cells in a monolayer without disrupting cells’ and vascular structures’ integrity. The contralateral femur was flushed with 500L of phosphate-buffered saline in the form of a fine cell suspension, centrifuged at 180g for 5 minutes, and the BM supernatant collected for further analysis.
Endothelial cell culture
The human umbilical vascular endothelial cell (HUVEC) line was cultured in complete EBM2 medium (10% fetal bovine serum, L-glutamine 1⫻, antimicotic-antibiotic 1⫻; Invitrogen) in 12-well 0.2% gelatin-coated plates and maintained at 37°C in a 5% CO2atmosphere. The cholesterol content of HUVEC monolayer was altered with low-density lipoprotein (LDL; 100g/mL), high-density lipoprotein (HDL; 100 g/mL), LDL together with low-density lipoprotein receptor (LDLR) blocking antibody (2g/mL) or HDL together with Blt3 (5M). After a 4-hour incubation period, cell-culture supernatants were collected for further analysis. Un-treated HUVECs were used as controls. Each experimental condition was performed in triplicate.
Biochemical measurements
Blood plasma was obtained by centrifugation of blood at 4°C and 1500g for 20 minutes and used for the determination of cholesterol and SDF-1 levels. Blood plasma cholesterol was measured in the Architect ci8200 analyzer (Abbott Diagnostics). Commercially available assays were used to measure SDF-1␣ levels in the blood plasma and in BM and cell culture supernatants (Quantikine; R&D Systems).
Immunohistochemistry and immunofluorescence
BM smears were air-dried, fixed in cold acetone for 10 minutes, and immunostained with a polyclonal rabbit anti–human factor VIII–related antigen antibody (Dako). Labeling was visualized with the use of the streptavidin-biotin-peroxidase method plus diaminobendizine (Dako), and counterstained with Mayer hematoxylin. Slides were scored by counting a total of 10 hot spots (in 2 to 3 slides for each animal) at a magnification of 100⫻ for MK counts. For immunofluorescence staining, BM smears were air-dried and fixed in methanol for 10 minutes at room temperature (RT), blocked with phosphate-buffered saline plus 0.1% bovine serum albumin for 45 minutes at RT, and incubated with primary antibody p-H3 (1:100, 06-570; Upstate Cell Signaling Solutions) at 4°C overnight. This antibody stains for the phosphorylated form of Histone 3, which occurs during prophase and is dephosphorylated during anaphase. The secondary anti-body used was anti–rabbit–coupled immunoglobulin G for 1 hour at RT (1:1000 Alexa Fluor 488; Molecular Probes). To assess cell apoptosis, analysis of DNA fragmentation by transferase-mediated dUTP nick-end labeling (TUNEL) was performed in BM smears with the use of In Situ Cell Death Detection kit, POD, and diaminobendizine substrate kits (Roche), following manufacturer’s instructions. In either staining, the percentage of positive cells was determined by counting a total of 500 cells/slide (400⫻ amplification) and by calculating the proportion of stained nuclei. For scoring, preference was given to areas presenting more intense and homogenous staining.
Flow cytometry
BM and PB mononuclear cells were stained for B lymphocytes, myeloid, endothelial, and progenitor cells by the use of antibodies against CD19 (1:100; R&D Systems), Gr1 (1:100; R&D Systems), Flk1 (1:400; R&D Systems), Sca1 (1:500; R&D Systems), and c-Kit (1:100; R&D Systems), respectively, for 1 hour at 4°C. Secondary antibodies used were anti–rat FITC/PE/APC-coupled immunoglobulin G (1:100, 30 minutes, 4°C Alexa
Fluor 488/594; Molecular Probes). In addition, cells were stained for CXCR4 (1:100; Chemicon) by use of the same protocol. Staining for Sca1, c-Kit, and CXCR4 was done after lineage-negative (Lin⫺) gating (Miltenyi Biotec). Flow cytometry was done in a FACSCalibur (BD Biosciences), and analysis was done with the use of CellQuest software.
Migration assay
Isolation of BM-derived B lymphocytes (CD19⫹) and progenitor cells (Lin⫺Sca1⫹c-Kit⫹) was performed with magnetic beads (Miltenyi Biotec) following the manufacturer’s instructions. For migration assays, cells (1⫻ 106/mL) were previously placed in serum-free medium for 4 hours in the presence or absence of LDL (100g/mL) or HDL (100 g/mL), alone or together with Blt3 (5M). Cell aliquots (100 L) were subsequently added to 5-m pore Transwell (Corning) inserts and plated into a 24-well plate. The lower compartment contained 600L of serum-free medium with or without SDF-1-␣ (100 ng/mL). The number of migrated cells was assessed after 6 hours by counting the cell number in 6 high-power fields (400⫻ magnification) with an optical microscope.
In vitro differentiation assay
Methylcellulose culture was performed following manufacturer’s instruc-tions (Methocult; StemCell Technologies) in a 24-well plate containing 1⫻ 104Lin⫺Sca1⫹c-Kit⫹cells per well and in the presence or absence of LDL (100g/mL) or HDL (100 g/mL) alone or together with Blt3 (5M). The number of colonies was scored after 7 days of culture with the use of an inverted microscope. Colony morphology was assessed on Giemsa-stained cytospins of selected colonies.
RNA extraction, cDNA synthesis, and quantitative reverse transcriptase polymerase chain reaction
BM cells were analyzed for LDLR, CD36, and SR-BI mRNA expression by quantitative reverse transcriptase polymerase chain reaction (RQT-PCR). Total cellular RNA was extracted, and cDNA was synthesized following conventional protocols. PCR was performed with a PCR thermal cycler (Uno II; Biometra). The primer sequences were as follows: LDLR forward primer: 5⬘-GGGCCTCTGTCTGGTGTTTA-3⬘; LDLR reverse primer: 5⬘-AGCAGGCTGGATGTCTCTGT-3⬘; CD36 forward primer: 5⬘-TGGAGCT-GTTATTGGTGCAG-3⬘; CD36 reverse primer: 5⬘-TGGGTTTTGCACAT-CAAAGA-3⬘; SR-BI forward primer: 5⬘-GGGCTCGATATTGATGGAGA-3⬘; and SR-BI reverse primer: 5⬘-GGAAGCATGTCTGGGAGGTA-3⬘. mRNA quantification was performed by the use of the ABI Prism 7700 Sequence Detection System and the SYBR Green Master Mix kit (both from Applied Biosystems).-Actin gene was used as standard reference (normalizer). The relative expression of each mRNA was calculated by use of the comparative threshold cycle method. Primer sequences for-actin were as follows: forward, 5⬘-AGCCATGTACGTAGCCATCC-3⬘; and reverse, 5⬘-CTCTCAGCTGTGGTGGTGAA-3⬘.
CXCR4 promoter activity (luciferase firefly reporter gene assay)
CXCR4 promoter was analyzed for transcription factor binding sites prediction by use of the TFBlast database (http://www.gene-regulation.com/cgi-bin/pub/ programs/tfblast/tfblat.cgi). Deletion fragments from promoters were amplified by PCR by use of the following primers 5⬘-3⬘: FOR1, ATGACGCGTCGAAG-GAAGGATCTTTACTCC; FOR2, ATGACGCGTCCT GGCATTTCATC-TCTCCG; and REV primer,ATGAAGCTTCCCCCAGCGCCACGACC;ACG.
MluI and SacI restriction sites (in bold) were included, respectively, in forward
and reverse primers. In a 24-well plate, 4⫻ 105cells per well were cultured in supplemented RPMI (10% fetal bovine serum, L-glutamine 1⫻, antimicotic-antibiotic 1⫻; Invitrogen). Transfections were performed with the use of Lipofectamine 2000 (Invitrogen) according to the standard protocol, 0.5g of the Renilla luciferase–expressing vector (transfection control), and 0.5g of the pGL3 construct of interest carrying the firefly luciferase. Twenty-four hours after transfection, cells were stimulated with LDL (100g/mL), HDL (100 g/mL), and SDF-1 (50 ng/mL) overnight at 37°C. The Dual-Luciferase Reporter Assay System (Promega Corporation) was used for the determination of CXCR4 promoter activity following the manufacturer’s instructions. Unstimulated cells
were used as controls. Results were corrected for transfection efficiency by the Renilla luciferase activity, and all conditions were performed in triplicate.
Statistical analysis
Results are expressed as mean plus or minus standard deviation. Data were analyzed by use of the unpaired 2-tailed Student t test. P values of less than .05 were considered significant.
Results
High-cholesterol diet induces elevated LDL cholesterol levels and inversion of the LDL to HDL cholesterol ratio
A 30-day high cholesterol feeding trial was conducted in C57Bl/6J mice, and total cholesterol, LDL, and HDL levels were assessed in the PB plasma. This high-cholesterol diet resulted in increased total cholesterol levels (Pⱕ .05; supplemental Figure 1A, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). LDL, 1 of the major 5 groups of lipoproteins and the major cell source of cholesterol, was elevated up to 8.5-fold in animals on high-cholesterol diet; and HDL, which is involved in cholesterol reverse transport, was elevated up to 1.8-fold (supple-mental Figure 1A). Increased LDL to HDL cholesterol ratio also was observed (Pⱕ .05; supplemental Figure 1B). The present model, also proposed by others as a murine model of hypercholes-terolemia,15 was seen here to be associated not only with high cholesterol and LDL blood plasma levels but also with inversion of the LDL to HDL cholesterol ratio.
A high-cholesterol diet is associated with thrombocytosis, lymphocytosis, and increased circulating progenitor cells
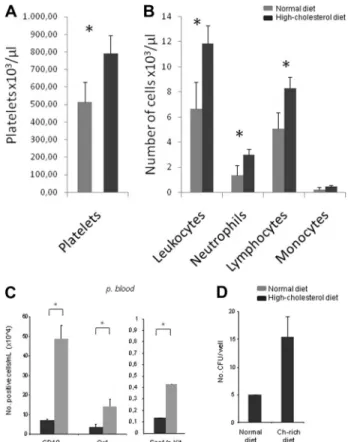
Complete blood cell count and leukocyte formula were performed in PB samples of mice fed a normal or high-cholesterol diet. Mice fed the high-cholesterol diet displayed a significant increase in platelet counts (thrombocytosis; Figure 1A), and leukocyte counts, specifically those of neutrophils and lymphocytes, also were increased (Pⱕ .05; Figure 1B). Detailed characterization of the circulating leukocytes through flow cytometry for Gr1, Sca1, c-Kit, and CD19 confirmed the increase in neutrophils (Gr1⫹), B lympho-cytes, and progenitor cells (Sca1⫹c-Kit⫹; Pⱕ .05; Figure 1C). Isolation of the mobilized Sca1⫹c-Kit⫹ cells from the PB con-firmed these are progenitor cells, as assessed by the generation of colony-forming units (CFUs) on methylcellulose (Figure 1D).
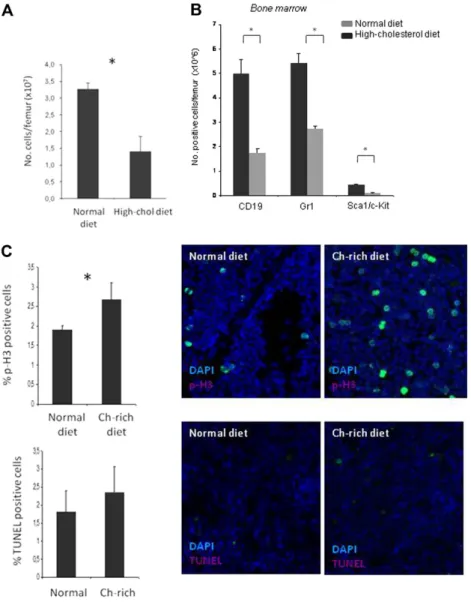
A high-cholesterol diet is associated with decreased total BM-cell counts and increased BM-cell proliferation
To determine whether the changes in the PB reflected those in the BM, absolute cell counts, proliferation, apoptosis, and cell distribu-tion pattern were assessed in the femur of mice fed normal and high-cholesterol diets. A decrease in total BM-cell counts was observed with the high-cholesterol diet (Pⱕ .05; Figure 2A), and, as determined by flow cytometry (for Gr-1, Sca1, c-Kit, and CD19), there was a major decrease in neutrophils (Gr-1⫹), progenitor cells (Sca1⫹c-Kit⫹), and B lymphocytes (CD19, Pⱕ .05; Figure 2B). No changes were seen in cell apoptosis, as assessed by TUNEL, but p-H3 staining of femur BM smears showed a significant increase in cell proliferation (Pⱕ .05) in the mice fed a high-cholesterol diet (Figure 2C). The decrease in absolute
BM-cell counts with a high-cholesterol diet was thus associated with an increase in BM-cell proliferation.
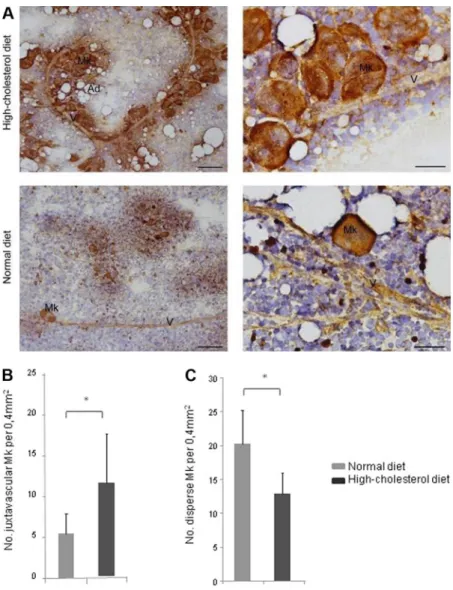
A high-cholesterol diet is associated with MK delocalization toward BM sinusoids
To evaluate whether thrombocytosis observed in mice fed a high-cholesterol diet was caused by increased MK numbers, cytologic evaluation of femur BM was performed in smears stained for factor VIII–related antigen (and also assessed by flow cytom-etry against CD41; data not shown). The total number of MKs in BM smears of normal and high-cholesterol diet mice was similar (supplemental Figure 2), but the MK distribution pattern was distinct. In the BM of mice fed a high-cholesterol diet, instead of occurring in small groups of cells, MKs formed large clusters in close contact with the BM sinusoids (Figure 3A). The number of MKs juxtaposed to the BM vasculature was therefore significantly greater in mice fed a high-cholesterol diet (Pⱕ .05; Figure 3B). Conversely, the number of MKs dispersed throughout the BM smear, nonassociated with the BM vasculature, was greater in mice fed the normal diet (Pⱕ .05; Figure 3C). Thrombocytosis, ob-served in mice fed the high-cholesterol diet, is therefore associated with massive MK delocalization toward BM vessels.
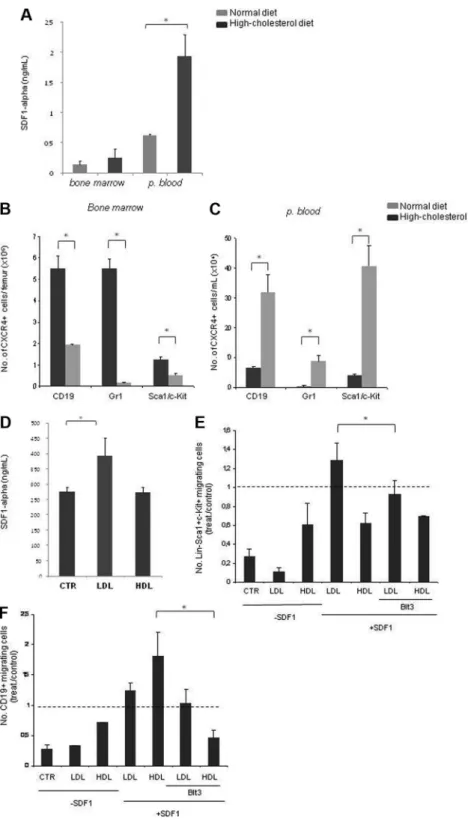
Figure 1. A high-cholesterol diet is associated with thrombocytosis, lymphocy-tosis, and increased circulating progenitor cells. (A) Hypercholesterolemia was accompanied by increased platelet counts (thrombocytosis;⫻103/L). (B) Besides thrombocytosis, leukocytosis also was observed in mice fed a high-cholesterol diet. The leukocytosis in mice fed a high-cholesterol diet was mainly caused by the significant increase in circulating lymphocytes and neutrophils (⫻103/L). (C) Flow cytometry analysis with Gr-1 (neutrophils), Sca1/c-Kit (progenitors), and CD19 (B lymphocytes) cell-surface markers confirms that the leukocytosis is mainly attributable to a massive increase in circulating lymphocytes (lymphocytosis) and neutrophils (neutrophilia;⫻103/L). In addition, hypercholesterolemia was also accompanied by an increase in the number of circulating progenitor cells (⫻104/mL; *P⬍ .05). (D) Isolated Lin⫺Sca1⫹c-Kit⫹cells from the PB of mice fed a normal diet and a high-cholesterol diet form CFUs in methylcellulose cultures, demonstrating their progenitor potential (*P⬍ .05). These experiments were performed 3 times with groups of 6 mice/experimental condition with consistent results.
SR-BI is abundantly expressed by BM cells
Next, to determine via which receptors cholesterol (namely LDL) exerted its effects in the BM microenvironment, we screened BM cells for mRNA expression of SR-BI, LDL receptor, and CD36 by QRT-PCR. SR-BI is highly expressed by neutrophils (Gr1⫹), B lymphocytes (CD19⫹), and progenitor cells (Sca1⫹; Figure 4), whereas BM endothelial cells express the 3 cholesterol receptors SR-BI, LDLR, and CD36. We used this information to test the direct and the indirect effects of cholesterol on HCs.
LDL does not promote cell proliferation but affects hematopoietic differentiation in vitro
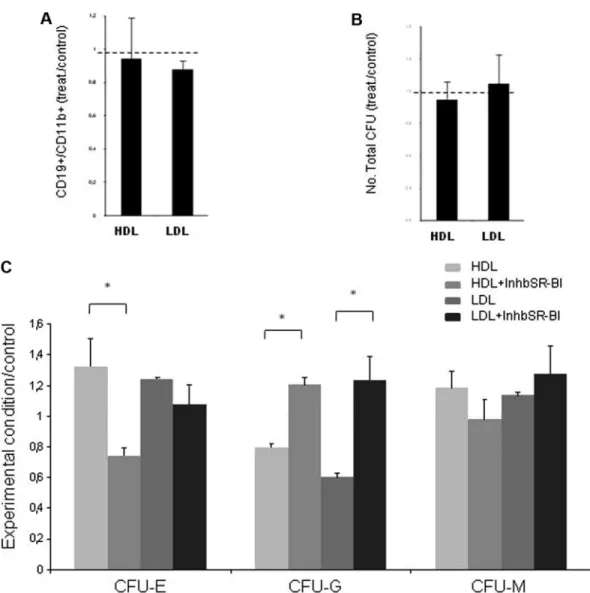
In vitro studies to test the direct effects of cholesterol on HCs were achieved through LDL and HDL exposure. To assess whether any of these lipoproteins conferred any proliferation advantage, the number of viable BM mononuclear cells, determined by Trypan blue exclusion and after LDL and HDL exposure, was compared with nontreated controls (supplemental Figure 3); no differences were registered.
Upon LDL and HDL exposure, lineage commitment from HPCs (cord blood–derived CD34⫹) in a coculture system with immortal-ized stromal cells (S17) showed no differences in myeloid versus
lymphoid commitment (assessed by flow cytometry for CD19 and CD11b), compared with untreated controls (Figure 5A). However, myeloid commitment quantified by clonogenic assays (in methyl-cellulose) was affected by LDL and HDL exposure. Although no differences in total CFUs were seen (Figure 5B), after 7 days of methylcellulose-based culture both LDL and HDL slightly in-creased the number of erythroid and macrophage CFU (ie, CFU-E and CFU-M), and significantly decreased that of granulocytic CFU (CFU-G; Figure 5C). These effects were reverted in the presence of the SR-BI inhibitor (Figure 6C).
A high-cholesterol diet induces increased SDF-1 plasma levels and disrupts the SDF-1:CXCR4 BM axis, favoring CXCR4ⴙcell mobilization to PB
Next we exploited the indirect effects of cholesterol exposure in mediating the exit of BM cells to the PB. We reasoned the BM-cell migration to the PB, observed with a high-cholesterol diet, could be attributable to changes in the levels of specific chemotactic cytokines, induced by hypercholesterolemia. We assessed SDF-1 PB plasma levels by enzyme-linked immunosorbent assay (ELISA) and found that these were elevated up to 3-fold in animals fed a high-cholesterol diet (Pⱕ .05; Figure 6A). To determine whether Figure 2. A high-cholesterol diet is associated with
de-creased total BM-cell counts. (A) Hypercholesterolemic mice present reduced cell numbers per femur (⫻107). (B) Flow cytometric analysis with Gr-1 (neutrophils), Sca1/c-Kit (progenitors), and CD19 (B lymphocytes) cell-surface mark-ers shows reduced numbmark-ers per femur (⫻106) of all cell lineages tested. (C) Hypercholesterolemia induces cell prolif-eration (p-H3 immunostaining, top) without altering cell apopto-sis (TUNEL assay, bottom; *P⬍ .05). These experiments were performed 3 times with groups of 6 mice/experimental condition with consistent results. Ch indicates cholesterol.
this elevation was accompanied by increased cell migration to the PB, double-positive CD19/CXCR4, Gr-1/CXCR4, and triple-positive Sca1/c-Kit/CXCR4 cells were quantified by flow cytom-etry in the BM and PB. A significant decrease in the number of double-positive cells in the BM was seen for B cells (CD19⫹CXCR4⫹) and progenitor cells (Sca1⫹c-kit⫹CXCR4⫹, Pⱕ .05; Figure 6B); in turn, B cells, neutrophils, and progenitor cells were significantly increased in the PB (Pⱕ .05; Figure 6C).
Moreover, as determined by ELISA, LDL treatment of endothe-lial cells (ie, HUVECs and also done with the use of BM endothelial cells [not shown]) significantly increased SDF-1 secre-tion (Pⱕ .05; Figure 6D). Taken together, these data show that elevated LDL induces SDF-1 secretion, resulting in elevated SDF-1 plasma levels and inducing CXCR4⫹cells exit into the PB.
LDL and HDL favor HC migration toward SDF-1 in vitro
A major effect of the high-cholesterol diet in vivo was the increased mobilization of lymphoid and HPCs from the BM to the PB; in vitro, no proliferation advantage or favoring of lymphoid over myeloid commit-ment was found with LDL/HDL enrichcommit-ment. Therefore, we reasoned the altered BM- and PB-cell counts could be attributable to a direct effect on cell mobilization/migration. After LDL and HDL exposure of BM B lymphocytes (CD19⫹) and progenitor cells (Lin⫺Sca1⫹c-Kit⫹), and considering SDF-1 alone as the control condition (and equal to 1), we found that LDL promoted progenitor cell and CD19⫹cell migration toward SDF-1 in Transwell migration assays, an effect that was reverted in the presence of SR-BI inhibitor (Pⱕ .05; Figure 6E-F). In contrast, HDL enrichment resulted in decreased progenitor cell migration (Figure 6E). LDL or HDL alone had a minor effect in either progenitor or CD19⫹cell migration. Neutrophils have been recently shown to migrate more efficiently toward KC, an ELR⫹chemokine,17and were therefore Figure 3. A high-cholesterol diet is associated with MK delocal-ization toward BM sinusoids. (A) Hypercholesterolemia is associ-ated with MK clustering on the BM vasculature lining (V), as detected by immunostaining for FVIIIra; bar, 100m. Greater magnification of BM smears clearly shows that MK are juxtaposed to the BM vascu-lature (V); bar, 30m. Ad, adipocytes. (B-C) Juxtavascular and dis-perse MKs and vessel count in BM smears, at low amplification, revealed that hypercholesterolemia is accompanied by an increase in juxtavascular and decrease in disperse MK. *P⬍ .05. These quanti-fications were obtained in 2 independent experiments in BM smears from 3 mice/experimental group with consistent results.
Figure 4. SR-BI is abundantly expressed by BM cells. QRT-PCR of cholesterol receptors mRNA expression screening on different BM-cell lineages demonstrates that SR-BI is the most frequent receptor expressed on the cell lineages tested, except for endothelial cells. In endothelial cells, CD36 had the greatest mRNA expression level. These experiments were obtained from 2 separate experiments, with consis-tent results.
not included in these experiments, in which we aimed at testing the effects of LDL on SDF-1–induced cell migration.
Cholesterol has minor effects on the CXCR4 promoter activity
Having shown that LDL promotes SDF-1 production by endothe-lial cells and promotes HC (B lymphoid, HPC) migration induced by SDF-1, next we determined whether LDL also regulated CXCR4 expression. By using leukemia cells lines representative of the hematopoietic lineages mobilized by high cholesterol (specifi-cally the 697 B leukemia cells, HEL erythroid/MK leukemia cells, and HL 60 monocytic leukemia cells), we observed that LDL does not significantly affect CXCR4 promoter activity on lymphoid or monocytic cells but reduces it on MK cells (supplemental Figure 4); interestingly, we observed that SDF-1 strongly induced CXCR4 promoter activity (data not shown).
Discussion
We show here that cholesterol exerts direct and indirect effects in the BM microenvironment, regulating HC retention versus
mobili-zation. Our data may be used to explain the hematopoietic phenotype of patients with hypercholesterolemia.
The mechanisms that regulate HPCs from the BM to the PB have been extensively studied. The current model put forward to explain the mechanisms by which different agents promote HPC mobilization suggests that, in steady state, HPCs are retained in the BM by SDF-1:CXCR4 interactions; conversely, perturbing the SDF-1:CXCR4 axis, either by increasing SDF-1 levels in the PB or by the use of CXCR4 antagonists, results in HPC mobilization.18 Other agents, namely the widely used granulocyte colony-stimulating factor, contribute toward HPC mobilization by induc-ing proteases activity in the BM microenvironment18-22and/or by altering (reducing) the SDF-1:CXCR4 interaction.23Other studies have suggested adhesion molecules24 and components of the complement pathway25,26may, directly or also by interfering with the SDF-1:CXCR4 signaling axis, contribute toward optimizing HPC mobilization.
Besides SDF-1 and CXCR4, the authors of other studies28-30 have focused on the importance of signals that regulate the integrity and response of the BM endothelial compartment in the mainte-nance of BM homeostasis, BM recovery after myeloablation, and Figure 5. LDL does not promote cell proliferation but affects hematopoietic differentiation in vitro. (A) B lymphoid-cell commitment is not altered by LDL (100g/mL) or HDL (100g/mL) exposure, defined as the ratio of CD19⫹(B lymphocytes) to CD11b⫹(myeloid) cells and assessed by flow cytometry. (B) LDL (100g/mL) or HDL (100g/mL) exposure does not change the total number of colonies formed in methylcellulose, a measure of CFUs. (C) LDL (100 g/mL) and HDL (100 g/mL) exposure increase the number of erythroid and macrophage CFU (CFU-E and CFU-M, respectively) and significantly decreases the number of granulocytic CFU (CFU-G). Inhibiting the cholesterol receptor SR-BI reverses the effect of LDL (*P⬍ .05). These data were obtained from 2 separate experiments with consistent results.
also in HPC mobilization. In this context, it has been shown that vascular endothelial growth factor (VEGF):VEGF receptor signal-ing pathway is an essential regulator of BM endothelium integrity and function. VEGF signaling in the BM microenvironment may also promote HPC mobilization in an indirect manner by affecting BM capillary leakiness.29-31
Interestingly, the same pathways (SDF-1, VEGF) that regulate HPC retention or mobilization from the BM microenvironment, and the integrity of BM endothelial cells, also regulate MK survival, proliferation, migration, and platelet production.13,32-35
Taken together, it has become increasingly clear that the cytokine milieu is crucial in the regulation of BM-cell retention versus mobilization. Much less is known about the effects of systemic stimuli, other than cytokines, in the BM microenviron-ment. In the present study, we hypothesized that cholesterol, known to affect systemic endothelial functions36-38 and also to affect monocyte/macrophage activation,6,39-43 might induce changes in the BM microenvironment.
The link between hypercholesterolemia (high systemic choles-terol levels) and hematopoiesis has been previously suggested. Figure 6. A high-cholesterol diet induces increased SDF-1 plasma levels, favors CXCR4ⴙcell mobiliza-tion to PB, and favors HC migramobiliza-tion toward SDF1. (A) Hypercholesterolemia is accompanied by an increase in PB plasma SDF-1 levels, as determined by ELISA quantification. (B) Flow cytometry analysis using Sca1/c-Kit (progenitor), CD19 (lymphocyte), and Gr-1 (neutro-phils) cell-surface markers together with CXCR4 shows reduced numbers of double-positive cells per femur (⫻106) for all cell lineages tested. (C) Flow cytometry analysis with Lin⫺Sca1⫹c-Kit⫹(progenitor), CD19⫹ (lym-phocyte), and Gr-1⫹(neutrophil) cell-surface markers together with CXCR4 shows increased numbers of double-positive B lymphocytes, neutrophils, and progeni-tor cells (⫻ 104) in the PB of high-cholesterol mice. (D) LDL exposure (100g/mL) increased SDF-1 produc-tion by HUVEC in vitro, as determined by ELISA (*P⬍ .05). These data were obtained from 3 separate experiments in which we used 6 mice per experimental condition with consistent results. (E) LDL (100g/mL) induces and HDL (100g/mL) reduces progenitor cells (Lin⫺Sca1⫹c-Kit⫹) migration toward SDF-1. (F) LDL (100g/mL) induced B-lymphocyte (CD19⫹) migration toward SDF-1 is reversed when SR-BI is inhibited. LDL effect is reverted when SR-BI is inhibited (*P⬍ .05). The data are shown as the number of migrating cells in relation to the control condition (SDF-1 alone). These data were obtained from 2 separate experiments with consistent results. CTR, control.
High cholesterol increases macrophage recruitment/mobilization and activation in atherosclerosis, for instance.5,6,40 Moreover, patients with hypercholesterolemia present with thrombocytosis and elevated leukocyte counts, specifically of lymphocytes and neutrophils.44The mechanistic basis for the hematopoietic effects of cholesterol, namely its effects in the BM microenvironment, has not been established.
We developed a model of murine hypercholesterolemia induced by a high-cholesterol diet, after which mice present elevated total cholesterol levels and a HDL-LDL inversion. Similar to patients with hypercholesterolemia, high-cholesterol mice present with increased lymphocytes (lymphocytosis), neutrophils (neutrophilia) and thrombocytosis, and also increased mobilization of HPCs.
A detailed analysis of the BM of normal-diet and high-cholesterol diet mice revealed that MK numbers were similar in both groups but that there was a massive accumulation of MKs around BM vessels in the latter. It is now clearly established that the interaction between MKs and the BM vessels is essential for platelet production and release.45,46Our observation of MK accumu-lation in close contact with BM vessels in high-cholesterol mice strongly suggests massive MK delocalization (migration) toward the BM vessels. As mentioned previously, SDF-1 is known to regulate MK migration and platelet production.12,13
Besides the massive accumulation of MKs around BM vessels, we also observed a significant increase in proliferating cells in the BM of high-cholesterol mice. Histologic analysis suggests these may be lymphocytes (data not shown). Elevated HDL or LDL did not affect proliferation of lymphoid versus myeloid commitment but significantly inhibited CFU-G differentiation in vitro. This inhibition may explain the reported decrease in monocytes in hypercholesterolemia patients.44
LDL increased SDF-1 production by endothelial cells in culture. Accordingly, mice fed the high-cholesterol diet had significantly increased PB SDF-1 plasma levels. We suggest this increased PB SDF-1 creates a perturbation in the SDF-1:CXCR4 axis, as evidenced by the decrease in CXCR4⫹B lymphocytes and of Lin⫺sca1⫹c-Kit⫹CXCR4⫹hematopoietic precursors in the BM of high-cholesterol mice, with a corresponding increase of these cell populations, and also of Gr-1⫹CXCR4⫹neutrophils, in the PB. Although neutrophils have been shown to migrate robustly to chemokines other than SDF-1, the dramatic increase in CXCR4⫹Gr-1⫹neutrophils in high-cholesterol mice suggests these cells are also strongly mobilized from the BM in response to the elevated cholesterol levels. Taken together, we conclude that high cholesterol, specifically elevated LDL, promotes BM-cell
mobiliza-tion and platelet producmobiliza-tion by increasing SDF-1 PB levels, which perturbs the BM SDF-1:CXCR4 axis.
In addition, LDL also promoted the migration of B lymphocytes and HPC toward SDF-1 in vitro, an effect that appeared dependent on SR-BI (a cholesterol receptor expressed by various cell types in the BM). Mechanistically, we have recently discovered that the migration of B-cell leukemias toward VEGF and SDF-1 is strongly promoted by cholesterol, via complement (specifically C3a) induc-tion (Fragoso R, T.C., A.L.G., S.D., manuscript in preparainduc-tion). Therefore, in the present report we suggest LDL may promote B cells and HPC migration toward SDF-1 via complement induc-tion. However, because CXCR4 function is dependent on its recruitment into lipid rafts,47 which in turn require membrane cholesterol, other mechanisms may be involved in the chemotactic-promoting role of LDL toward SDF-1, including lipid rafts formation or mobility.
The data obtained from our mouse model of hypercholesterol-emia strongly suggest that elevated cholesterol levels and specifi-cally elevated LDL interfere with the SDF-1:CXCR4 axis in the BM microenvironment and affect BM homeostasis. These findings have implications for clinical studies aimed at optimizing HC mobilization for therapeutic purposes and may also be important in the context of BM malignancies.
Acknowledgments
We thank other members of the Angiogenesis laboratory for their suggestions.
T.C. and J.S. are recipients of fellowships from Fundac¸a˜o para a Ciencia e a Tecnologia (FCT, Portuguese Government). This study was supported by GlaxoSmithKline.
Authorship
Contribution: S.S.D. designed the project; J.S. and C.T. performed the CXCR4 promoter experiments; T.C. performed the BM immu-nohistochemical staining; A.L.G. performed the in vivo and the in vitro experiments; and A.L.G. and S.D. wrote the paper.
Conflict-of-interest disclosure: The authors declare no compet-ing financial interests.
Correspondence: Se´rgio Dias, Angiogenesis Group, CIPM/ IPOLGF, Rua Professor Lima Basto, 1099-023 Lisboa, Portugal; e-mail: [email protected].
References
1. Kaplan RN, Psaila B, Lyden D. Niche-to-niche migration of bone-marrow-derived cells. Trends Mol Med. 2007;13(2):72-81.
2. Kopp HG, Avecilla ST, Hooper AT, et al. Tie2 acti-vation contributes to hemangiogenic regeneration after myelosuppression. Blood. 2005;106(2):505-513.
3. Steinberg D. Atherogenesis in perspective: hy-percholesterolemia and inflammation as partners in crime. Nat Med. 2002;8(11):1211-1217. 4. Owens D, Collins P, Johnson A, Tomkin G.
Cellu-lar cholesterol metabolism in mitogen-stimulated lymphocytes—requirement for de novo synthesis. Biochim Biophys Acta. 1990;1051(2):138-143. 5. Han KH, Han KO, Green SR, Quehenberger O.
Expression of the monocyte chemoattractant pro-tein-1 receptor CCR2 is increased in hypercho-lesterolemia. Differential effects of plasma
li-poproteins on monocyte function. J Lipid Res. 1999;40(6):1053-1063.
6. Bobryshev YV. Monocyte recruitment and foam cell formation in atherosclerosis. Micron. 2006; 37(3):208-222.
7. Pathansali R, Smith N, Bath P. Altered megakaryocyte-platelet haemostatic axis in hy-percholesterolaemia. Platelets. 2001;12(5):292-297.
8. Bath PM, Gladwin AM, Carden N, Martin JF. Megakaryocyte DNA content is increased in pa-tients with coronary artery atherosclerosis. Car-diovasc Res. 1994;28(9):1348-1352. 9. Ebbe S, Dalal K, Forte T, Tablin F. Microcytic
thrombocytosis, small megakaryocytes, platelet lipids and hyperreactivity to collagen, lymphocy-topenia, eosinophilia, and low blood volume in genetically hyperlipidemic rabbits. Exp Hematol. 1992;20(4):486-493.
10. Mazoyer EES, Dalal K, Leven R, et al. Morpho-logical and kinetic abnormalities of platelets in hypercholesterolemic rabbits. Atherosclerosis. 1988;74(1-2):23-32.
11. Christenson JT. Preoperative lipid-control with simvastatin reduces the risk of postoperative thrombocytosis and thrombotic complications fol-lowing CABG. Eur J Cardiothorac Surg. 1999; 15(4):394-400.
12. Lane WJ, Dias S, Hattori K, et al. Stromal-derived factor 1-induced megakaryocyte migration and platelet production is dependent on matrix metal-loproteinases. Blood. 2000;96(13):4152-4159. 13. Hamada T, Mohle R, Hesselgesser J, et al.
Trans-endothelial migration of megakaryocytes in re-sponse to stromal cell-derived factor 1 (SDF-1) enhances platelet formation. J Exp Med. 1998; 188(3):539-548.
14. Avecilla ST, Hattori K, Heissig B, et al. Chemo-kine-mediated interaction of hematopoietic pro-genitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med. 2004; 10(1):64-71.
15. Paigen B. Genetics of responsiveness to high-fat and high-cholesterol diets in the mouse. Am J Clin Nutr. 1995;62(2):458S-462S.
16. Shalaby F, Rossant J, Yamaguchi TP, et al. Fail-ure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376(6535): 62-66.
17. Pelus LM, Bian H, Fukuda S, Wong D, Merzouk A, Salari H. The CXCR4 agonist peptide, CTCE-0021, rapidly mobilizes polymorphonuclear neu-trophils and hematopoietic progenitor cells into peripheral blood and synergizes with granulocyte colony-stimulating factor. Exp Hematol. 2005; 33(3):295-307.
18. Le´vesque JP, Hendy J, Takamatsu Y, Simmons PJ, Bendall LJ. Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by GCSF or cyclo-phosphamide. J Clin Invest. 2003;111(2):187-196.
19. Pelus LM, Fukuda S. Chemokine-mobilized adult stem cells; defining a better hematopoietic graft. Leukemia. 2008;22(3):466-473.
20. Pelus LM, Fukuda S. Peripheral blood stem cell mobilization: the CXCR2 ligand GRObeta rapidly mobilizes hematopoietic stem cells with en-hanced engraftment properties. Exp Hematol. 2006;34(8):1010-1020.
21. Petit IS-KM, Nagler A, Lahav M, et al. G-CSF in-duces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002;3(7):687-694.
22. Wengner AM, Pitchford SC, Furze RC, Rankin SM. The coordinated action of G-CSF and ELR⫹ CXC chemokines in neutrophil mobilization dur-ing acute inflammation. Blood. 2008;111(1):42-49.
23. Christopher MJ, Liu F, Hilton MJ, Long F, Link DC. Suppression of CXCL12 production by bone mar-row osteoblasts is a common and critical pathway for cytokine-induced mobilization. Blood. 2009; 114(7):1331-1339.
24. Ramirez P, Rettig MP, Uy GL, et al. BIO5192, a small molecule inhibitor of VLA-4, mobilizes
he-matopoietic stem and progenitor cells. Blood. 2009;114(7):1340-1343.
25. Reca R, Wysoczynski M, Yan J, Lambris JD, Ratajczak MZ. The role of third complement com-ponent (C3) in homing of hematopoietic stem/ progenitor cells into bone marrow. Adv Exp Med Biol. 2006;586:35-51.
26. Ratajczak J, Reca R, Kucia M, et al. Mobilization studies in mice deficient in either C3 or C3a re-ceptor (C3aR) reveal a novel role for complement in retention of hematopoietic stem/progenitor cells in bone marrow. Blood. 2004;103(6):2071-2078.
27. Carmeliet P, Ferreira V, Breier G, et al. Abnormal blood vessel development and lethality in em-bryos lacking a single VEGF allele. Nature. 1996; 380(6573):435-439.
28. Hooper AT, Butler JM, Nolan DJ, et al. Engraft-ment and reconstitution of hematopoiesis is de-pendent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell. 2009; 4(3):263-274.
29. Hattori KDS, Heissig B, Hackett NR, et al. Vascu-lar endothelial growth factor and angiopoietin-1 stimulate postnatal hematopoiesis by recruitment of vasculogenic and hematopoietic stem cells. J Exp Med. 2001;193(9):1005-1014.
30. Pitchford SC, Furze RC, Jones CP, Wengner AM, Rankin SM. Differential mobilization of subsets of progenitor cells from the bone marrow. Cell Stem Cell. 2009;4(1):62-72.
31. Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for di-agnosis and therapy. J Clin Oncol. 2002;20(21): 4368-4380.
32. Casella I, Feccia T, Chelucci C, et al. Autocrine-paracrine VEGF loops potentiate the maturation of megakaryocytic precursors through Flt1 recep-tor. Blood. 2003;101(4):1316-1323.
33. Majka M, Janowska-Wieczorek A, Ratajczak J, et al. Stromal-derived factor 1 and thrombopoietin regulate distinct aspects of human megakaryo-poiesis. Blood. 2000;96(13):4142-4151. 34. Mo¨hle R, Green D, Moore MA, Nachman RL,
Rafii S. Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc Natl Acad Sci U S A. 1997;94(2):663-668.
35. Junt T, Schulze H, Chen Z, et al. Dynamic visual-ization of thrombopoiesis within bone marrow. Science. 2007;317(5845):1767-1770. 36. Dart AM, Chin-Dusting JP. Lipids and the
endo-thelium. Cardiovasc Res. 1999;43(2):308-322. 37. Sima AV, Stancu CS, Simionescu M. Vascular
endothelium in atherosclerosis. Cell Tissue Res. 2009;335(1):191-203.
38. Wilkinson IB, Cockcroft JR. Cholesterol, endothe-lial function and cardiovascular disease. Curr Opin Lipidol. 1998;9(3):237-242.
39. Linton MF, Fazio S. Macrophages, inflammation, and atherosclerosis. Int J Obes Relat Metab Dis-ord. 2003;27(suppl 3):S35-S40.
40. Namiki M, Kawashima S, Yamashita T, et al. Lo-cal overexpression of monocyte chemoattractant protein-1 at vessel wall induces infiltration of mac-rophages and formation of atherosclerotic lesion: synergism with hypercholesterolemia. Arterio-scler Thromb Vasc Biol. 2002;22(1):115-120. 41. Shibata N, Glass CK. Regulation of macrophage
function in inflammation and atherosclerosis. J Lipid Res. 2009;50(suppl):S277-S281. 42. Takeya M. Monocytes and
macrophages—multi-faced cell population involved in inflammation, atherosclerosis, and obesity. Nippon Rinsho. 2005;63(suppl 4):117-122.
43. Watanabe T, Fan J. Atherosclerosis and inflam-mation mononuclear cell recruitment and adhe-sion molecules with reference to the implication of ICAM-1/LFA-1 pathway in atherogenesis. Int J Cardiol. 1998;66(suppl 1):S45-S53; discussion S55.
44. Huang ZS, Wang CH, Yip PK, Yang CY, Lee TK. In hypercholesterolemia, lower peripheral mono-cyte count is unique among the major predictors of atherosclerosis. Arterioscler Thromb Vasc Biol. 1996;16(2):256-261.
45. Dhanjal TS, Pendaries C, Ross EA, et al. A novel role for PECAM-1 in megakaryocytokinesis and recovery of platelet counts in thrombocytopenic mice. Blood. 2007;109(10):4237-4244. 46. Kopp HG, Rafii S. Thrombopoietic cells and the
bone marrow vascular niche. Ann N Y Acad Sci. 2007;1106:175-179.
47. Nguyen DHTD. CXCR4 function requires mem-brane cholesterol: implications for HIV infection. J Immunol. 2002;168(8):4121-4126.