Brasil
Carlos Eduardo Frigério Domingues
INSTITUTO DE BIOCIÊNCIAS DE BOTUCATU
Análise de ligação e associação no genoma com gagueira
desenvolvimental persistente em famílias do Estado de São Paulo
Brasil
Candidato: Carlos Eduardo Frigério Domingues Orientador: Prof. Dr. Danilo Moretti-Ferreira Co-orientador: Dennis Drayna, PhD
Tese apresentada ao Instituto de Biociências, Câmpus de Botucatu, UNESP, para obtenção do título de Doutor no Programa de Pós-Graduação em Ciências Biológicas Genética.
Análise de ligação e associação no genoma com gagueira desenvolvimental persistente em famílias do Estado de São Paulo – Brasil / Carlos Eduardo Frigério Domingues. – Botucatu : [s.n.], 2013
Tese (doutorado) - Universidade Estadual Paulista, Instituto de Biociências de Botucatu
Orientador: Danilo Moretti-Ferreira Coorientador: Dennis T. Drayna Capes: 40101002
1. Genética humana. 2. Distúrbios da fala. 3. Gagueira – Aspectos genéticos. 4. Cromossomos humanos – Anomalias. 5. Genes.
Palavras-chave: Análise de ligação; Cromossomo; 10q; Gagueira; GNPTAB;
Perdoe-as assim mesmo.
Se você é gentil,
as pessoas podem acusá-lo de interesseiro.
Seja gentil assim mesmo.
Se você é um vencedor,
terá alguns falsos amigos e alguns inimigos verdadeiros.
Vença assim mesmo.
Se você é honesto e franco,
as pessoas podem enganá-lo.
Seja honesto e franco assim mesmo.
O que você levou anos para construir,
alguém pode destruir de uma hora para outra.
Construa assim mesmo.
Se você tem paz e é feliz,
as pessoas podem sentir inveja.
Seja feliz assim mesmo.
O bem que você faz hoje
pode ser esquecido amanhã.
Faça o bem assim mesmo.
Dê ao mundo o melhor de você,
mas isso pode não ser o bastante.
Dê o melhor de você assim mesmo.
Veja você que, no final das contas,
é tudo entre você e Deus.
Nunca entre você e os outros.”
Dedico este trabalho...
À DEUS, pelo seu amor incondicional, por ser o princípio e o fim de tudo, e por estar sempre presente e pronto para ouvir, perdoar e acolher. Obrigado Senhor, por estar sempre comigo nos momentos de fé e também nas horas mais difíceis.
Aos meus pais, Antonio e Maria, por terem me dado a vida e por sempre acreditarem em mim. Há 34 anos, vocês insistentemente tem me apresentado exemplos de Amor, Dedicação, Honestidade, Paciência, Renuncia, Fé....que jamais esquecerei!
À minha sempre irmãzinha, Ana Paula. Minha amiga, minha companheira de luta. Obrigado pelo seu amor e carinho!
À minha Patrícia (e família), minha companheira de caminhada, minha companheira de vida. Obrigado pelo apoio, carinho, paciência e amor. Obrigado pela compreensão, por estar sempre comigo mesmo nos momentos mais distantes e difíceis. Obrigado por me ouvir, por me entender e por me dar força para a conclusão deste trabalho!
Aos portadores de gagueira, pacientes e familiares e, a todos os voluntários que colaboraram com este estudo. Obrigado por ainda acreditarem no valor da Educação,
Botucatu e a todo corpo docente e funcionários.
Ao National Institutes of Health – NIH, Bethesda, MD – USA e ao Graduate Partnerships Program (GPP), pela recepção, apoio e suporte.
Ao Instituto de Biociências de Botucatu, sua direção, corpo docente, funcionários e alunos.
Ao National Institute on Deafness and Other Communication Disorders – NIDCD, Rockville, MD – USA, funcionários, colaboradores, pesquisadores e alunos.
Ao Departamento de Genética, docentes e funcionários, por disponibilizar a infraestrutura para a realização deste trabalho.
Ao Departamento de Fonoaudiologia e ao Centro de Estudos da Educação e da Saúde de Marília (CEES/Unesp/Marília) por toda a infraestrutura e recrutamento dos pacientes. Ao Departamento de Fonoaudiologia, Fisioterapia e Terapia Ocupacional da Faculdade de Medicina da Universidade de São Paulo (FOFITO/FMUSP/São Paulo) por toda a infraestrutura e recrutamento dos pacientes.
À Stuttering Foundation of America, the National Stuttering Association, USA, e ao Hollins Communications Research Institute, pelo recrutamento de pacientes.
Ao Programa de Pós-graduação em Ciências Biológicas: área de concentração Genética e à Seção de pós-graduação (funcionários) que sempre atendeu minhas solicitações prontamente.
Ao Serviço de Aconselhamento Genético e todos os seus funcionários e colaboradores. À Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), pela bolsa concedida.
Ao meu orientador Prof. Adj. Danilo Moretti-Ferreira, pelos ensinamentos, amizade, paciência e confiança para o desenvolvimento deste trabalho.
Ao meu co-orientador Dr. Dennis Drayna, pessoa de singular capacidade intelectual e científica. Sou eternamente grato pela oportunidade e por ter acreditado em mim para o desenvolvimento desse trabalho em seu laboratório sob sua orientação e também pelo suporte financeiro para esse projeto (NIH GRANT NUMBER Z01-000046-12).
À Profa. Dra. Célia Maria Giacheti, pelo apoio e colaboração prestados nesse projeto. À Profa. Dra. Claudia Regina Furquim de Andrade, pelo apoio e colaboração prestados nesse projeto.
À Profa. Dra. Fabíola Staróbole Just, pelo auxílo e colaboração com os multirões na clínica de fonoaudiologia para o recrutamento de pacientes.
Ao Dr. Gustavo Henrique Vieira, meu grande amigo, companheiro que esteve comigo ao longo de todos esses anos de formação acadêmica. Aos inúmeros momentos que sempre esteve disposto a me ajudar, a me ouvir, a discutir enfim....e aos seus generosos préstimos viajando, com seu próprio carro para diversas cidades em busca de pacientes. Aos meus amigos Bruno Faulin Gamba, Marcos Leite Santono, Bianca Santos Domingues, Maria Alice, Breila Vilela de Oliveira pelo imenso e impagável apoio que sempre me deram ajudando nas coletas, emprestando seus carros para as viagens, pela ajuda na organização dos dados e no processamento das amostras, pelas conversas enfim por tudo.
Aos meus três grandes amigos que fiz nos Estados Unidos da América e que me acolheram num dos momentos mais delicados da minha vida. Obrigado pela amizade, pelo carinho que me receberam e por me mostrarem um pouco deste grandioso país, Mary Vandenbroucke, Eduardo Sainz e sua esposa Karen (in memorian).
Aos meus amigos de laboratório Dr. Changsoo Kang, Dr. Muhammad Hashim Raza, Dr. Tae-Un Han e a Emly Paris pela recepção, carinho, apoio, ensinamentos, conversas, ajuda nos experimentos, paciência....sem você não teria aprendido e conseguido ter feito tudo que fiz.
Aos meus colegas,vizinhos de laboratório, Dr. Kiyoto Kurima, Dra. Parna Chattaraj, Dr. Hiroshi Nakanishi, Dr. Taku Ito pela amizade e consideração.
Á Li Chunqin e ao Dr. Robert J. Morell pela viabilização dos procedimentos laboratoriais e obtenção dos resultados.
Aos meus amigos Uilian de Andreis, Marina Trevizan Guerra, Jossimara Poletini pelo carinho, pelas conversas, amizade e apoio.
LISTA DE FIGURAS...5
RESUMO...7
ABSTRACT ...8
1. INTRODUÇÃO ...9
1.1. DEFINIÇÃO DE GAGUEIRA ... 9
1.2. CLASSIFICAÇÃO DA GAGUEIRA ... 11
1.3. DIAGNÓSTICO DA GAGUEIRA ... 12
2. REVISÃO DA LITERATURA ...14
2.1. INCIDÊNCIA E PREVALÊNCIA ... 14
2.2. ETIOLOGIA ... 15
2.3. FATORES GENÉTICOS ... 18
2.4. DOENÇAS COMPLEXAS ... 20
2.4.1. Estratégias de análise ... 20
2.4.1.1. Análise de ligação paramétrica ... 24
2.4.1.2. Análise de ligação não paramétrica ... 25
2.4.1.3. Estudos de associação ... 25
2.5. ANÁLISES DE LIGAÇÃO E A GAGUEIRA ... 26
2.6. ESTUDO DE GENES CANDIDATOS ... 30
2.6.1. Genes candidatos e a gagueira ... 31
2.6.2. Genes FOXP2 e CNTNAP2 ... 36
2.6.2.1. Gene FOXP2 ... 36
2.6.2.2. Gene CNTNAP2 ... 37
2.7. POPULAÇÃO BRASILEIRA ... 38
3. OBJETIVOS ...40
3.1. PRIMÁRIOS ... 40
3.2. SECUNDÁRIOS ... 40
4. MATERIAL E MÉTODOS ...41
4.1. CASUÍSTICA ... 41
4.1.1. Grupo amostral ... 41
4.1.1.1. Critérios de inclusão ... 41
4.1.2. Grupo Controle ... 42
4.1.2.1. Critérios de inclusão ... 42
4.1.2.2. Critérios de exclusão ... 42
4.2. DIAGNÓSTICO DA GAGUEIRA ... 43
4.3. ASPECTOS ÉTICOS ... 44
4.4. COLETA DE AMOSTRAS BIOLÓGICAS ... 44
4.5. OBTENÇÃO DO DNA GENÔMICO ... 45
4.5.1. Quantificação, qualidade e integridade das amostras de DNA genômico ... 45
4.6. PROCEDIMENTOS E ANÁLISES MOLECULARES ... 46
4.6.1. Genotipagem ... 46
4.6.1.1. Marcadores SNP ... 46
4.6.1.2. Marcadores microssatélites ... 49
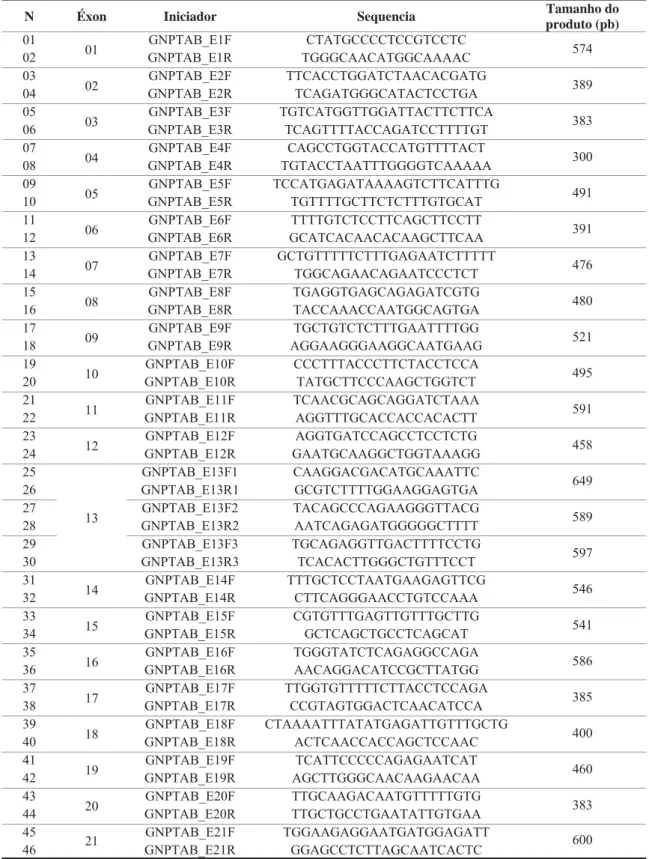
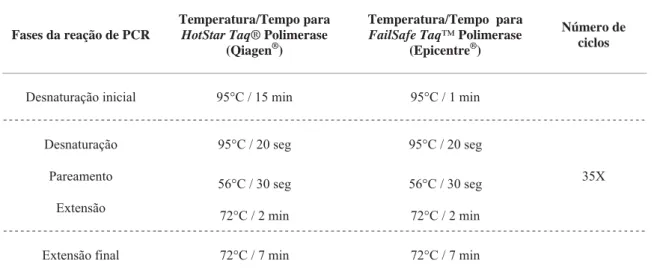
4.6.2. Sequenciamento do DNA ... 51
4.6.2.1. Desenho dos iniciadores ... 51
4.6.2.2. Análise dos éxons ... 56
4.6.3. Sequenciamento de Nova Geração (NGS, Next Generation Sequencing) ... 59
4.6.3.1. Plataforma SOLiD™ (Sequencing by Oligonucleotide Ligation and Detection) – Sequenciamento Exômico Completo (Whole exome sequencing) .. 60
4.6.4. Análises estatísticas ... 64
4.6.4.1. Programa easyLinkage ... 64
4.6.4.2. Plataforma Computacional Helix (NIH Helix Systems) ... 64
4.6.4.3. Procedimentos gerais ... 65
5. RESULTADOS ...66
5.1. GRUPO AMOSTRAL ... 66
5.2. ARTIGOS CIENTÍFICOS ... 77
5.2.1. Artigo científico 01 ... 77
5.2.2. Artigo científico 02 ... 80
5.3. DADOS - SEQUENCIAMENTO DO DNA ... 95
5.4. DADOS - SEQUENCIAMENTO DE NOVA GERAÇÃO ... 102
6. DISCUSSÃO E CONCLUSÃO ...106
7. REFERÊNCIAS BIBLIOGRÁFICAS ...111
ANEXO A ...132
ANEXO B ...133
ANEXO D ...134
ANEXO E ...139
APÊNDICE A ...141
APÊNDICE B...144
APÊNDICE C ...145
APÊNDICE D ...146
APÊNDICE E...148
APÊNDICE F ...150
APÊNDICE G ...152
LISTA DE TABELAS
Tabela 1. Variações nas estimativas de prevalência em diferentes estudos e populações.
... 15
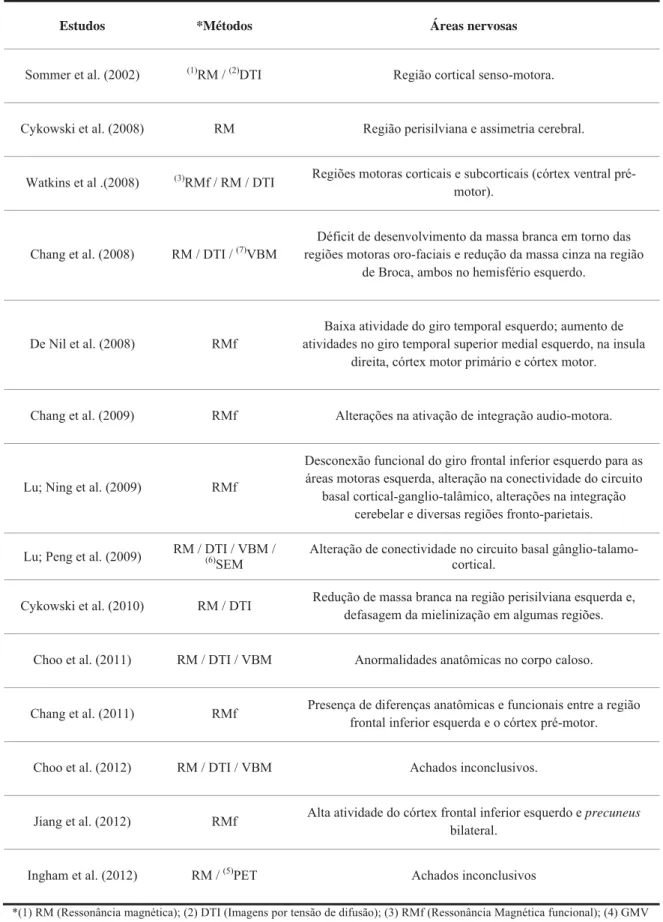
Tabela 2. Estudos de neuroimagem e a identificação de possíveis regiões relacionadas à gagueira desenvolvimental persistente. ... 17
Tabela 3. Estudos de ligação desenvolvidos em famílias portadoras de gagueira desenvolvimental persistente. ... 30
Tabela 4. Programa utilizado nas reações de PCR para marcadores microssatélites. ... 50
Tabela 5. Informações referentes ao gene GNPTAB e os iniciadores utilizados. ... 53
Tabela 6. Informações referentes ao gene GNPTG e os iniciadores utilizados. ... 54
Tabela 7. Informações referentes ao gene NAGPA e os iniciadores utilizados. ... 54
Tabela 8. Informações referentes ao gene DRD2 e os iniciadores utilizados. ... 54
Tabela 9. Informações referentes ao gene CNTNAP2 e os iniciadores utilizados. ... 55
Tabela 10. Informações referentes ao gene FOXP2 (isoforma II) e os iniciadores utilizados. ... 56
Tabela 11. Programas utilizados para as reações de amplificação por PCR. ... 57
Tabela 12. Programa utilizado para a reação de sequenciamento dos produtos de PCR. ... 58
Tabela 13. Distribuição do número de indivíduos, não relacionados, com gagueira desenvolvimental persistente familial e, casos controle. ... 66
Tabela 14. Distribuição do número de famílias com gagueira desenvolvimental persistente, não relacionadas. ... 67
Tabela 15. Relação de variações identificadas no gene GNPTAB. ... 96
Tabela 16. Relação de variações identificadas no gene GNPTG. ... 97
Tabela 17. Relação de variações identificadas no gene NAGPA ... 98
Tabela 18. Relação de variações identificadas no gene CNTNAP2 ... 99
Tabela 19. Relação de variações identificadas no gene FOXP2 ... 101
Tabela 20. Variações selecionadas após análise sistemática dos dados de sequenciamento de nova geração. ... 105
Tabela 21. Classificação da gravidade da gagueira segundo o teste SSI – 3 (Riley, 1994). ... 133
Tabela 22. Relação de iniciadores para os marcadores microssatélites selecionados e utilizados para o refinamento do mapa genético de ligação. ... 141
LISTA DE FIGURAS
Figura 1. Classificação da gagueira, modificado de Oliveira et al (2012). ... 12
Figura 2. Esquema com as etapas de análise para doenças comuns e complexas, modificado de Haines & Pericak-Vance (1998). ... 22
Figura 3. Esquema simplificado da via metabólica lisossomal (KANG; DRAYNA, 2012). ... 34
Figura 4. Esquema representando a sequência de etapas para a montagem da biblioteca genômica. ... 61
Figura 5. Heredogramas: BRPD01, BRPD02, BRPD03, BRPD04, BRPD05, BRPD06. ... 69
Figura 6. Heredogramas: BRPD07, BRPD08, BRPD09, BRPD10, BRPD11, BRPD12. ... 70
Figura 7. Heredogramas: BRPD13, BRPD14, BRPD15, BRPD16, BRPD17, BRPD18. ... 71
Figura 8. Heredogramas: BRPD19, BRPD20, BRPD21, BRPD22, BRPD23, BRPD24, BRPD25, BRPD26. ... 72
Figura 9. Heredogramas: BRPD27, BRPD28, BRPD29, BRPD30, BRPD31, BRPD33. ... 73
Figura 10. Heredogramas: BRPD34, BRPD35, BRPD38, BRPD45, BRPD47. ... 74
Figura 11. Heredogramas: BRPD48, BRPD49, BRPD50 ... 75
Figura 12. Heredogramas: BRPD51, BRPD52, BRPD53. ... 76
Figura 13. Esquema representando a sequência de etapas adotadas para a análise de dados obtidos pelo sequenciamento de nova geração. ... 103
Figura 14. Distribuição das variações, identificadas por WES, em cada família a partir do sequenciamento por eletroforese capilar. ... 104
Figura 15. Exemplo de eletroferogramas representando os alelos durante a análise de fragmentos (microssatélites) ... 144
Figura 16. Imagem da interface gráfica do Programa IGV (Integrative Genomics Viewer). ... 145
Figura 17. Exemplo de visualização gráfica parcial de uma região sequenciada por NGS (Next Generation Sequencing) – WES (Whole Exome Sequencing) após seleção do cromossomo e zoom ... 145
Figura 18. Dados referentes à análise sob modelo de herança dominante - BRPD47. 148 Figura 19. Dados referentes à análise sob modelo de herança recessivo - BRPD47 .. 149
Figura 20. Dados referentes à análise sob modelo de herança dominante - BRPD50 150 Figura 21. Dados referentes à análise sob modelo de herança recessivo - BRPD50 .. 151
Figura 22. Dados referentes à análise combinada, sob modelo de herança dominante - BRPD47_50. ... 152
Figura 23. Gene GNPTAB, éxon 02, sequenciamento bidirectional.. ... 153
Figura 24. Gene GNPTAB, éxon 04, sequenciamento bidirectional. ... 153
Figura 25. Gene GNPTAB, éxon 09, sequenciamento bidirectional. ... 154
Figura 26. Gene GNPTAB, éxon 10, sequenciamento bidirectional. ... 154
Figura 28. Gene GNPTG, éxon 6, sequenciamento bidirectional. ... 155
Figura 29. Gene GNPTG, éxon 7, sequenciamento bidirectional ... 156
Figura 30. Gene GNPTG, éxon 11, sequenciamento bidirectional. ... 156
Figura 31. Gene NAGPA, éxon 1, sequenciamento bidirectional... 157
Figura 32. Gene NAGPA, éxon 6, sequenciamento bidirectional... 157
Figura 33. Gene NAGPA, éxon 9, sequenciamento bidirectional... 158
Figura 34. Gene NAGPA, éxon 10, sequenciamento bidirectional... 158
Figura 35. Gene CNTNAP2, éxon 3, sequenciamento bidirectional. ... 159
Figura 36. Gene CNTNAP2, éxon 6, sequenciamento bidirectional. ... 159
Figura 37. Gene CNTNAP2, éxon 8, sequenciamento bidirectional. ... 160
Figura 38. Gene FOXP2, éxon 7, sequenciamento bidirectional. ... 160
Figura 39. Gene FOXP2, éxon 12, sequenciamento bidirectional. ... 161
RESUMO
A gagueira é uma doença comum que afeta a fluência da fala, caracterizada por repetições ou prolongamentos frequentes de sons, sílaba, palavras, ou por hesitações, ou interrupções no fluxo normal da fala. Trata-se de uma doença que tipicamente surge na infância em crianças com idade entre dois e quatro anos, com taxa de incidência estimada em torno de 5% da população. No entanto devido à elevada taxa de recuperação espontânea, estima-se uma prevalência de 1% na população em geral. Apesar do envolvimento de fatores ambientais, o fator genético é determinante para o desenvolvimento da doença. Algumas evidências que sustentam essa relação são: agregação familial, estudos com gêmeos e envolvendo adoções e, relações de consanguinidade em famílias com diversos afetados. Entretanto, trata-se de uma doença complexa na qual a identificação exata do tipo de herança é difícil de ser determinada uma vez que não seguem as leis de Mendel. Este estudo teve como principal finalidade realizar análises de ligação em 43 famílias brasileiras do Estado de São Paulo, não relacionadas, portadoras de gagueira desenvolvimental persistente a fim de identificar regiões cromossômicas com possíveis genes candidatos; Para as análises de ligação, inicialmente foi realizada a genotipagem de todas as famílias através de chips específicos para essa finalidade contendo 6056 marcadores SNP. Posteriormente, para o refinamento do mapa de ligação foram utilizados marcadores microssatélites polimórficos marcados com fluoróforos e analisados em sequenciador automático capilar. Para as estimativas das frequências alélicas destes marcadores na população brasileira foram utilizados amostras do grupo controle, não relacionadas, previamente selecionadas. Apenas duas famílias (BRPD_47 e BRPD_50) sob o padrão de herança dominante apresentaram significativas evidências de ligação na região 10q21, quando combinadas, LOD escore single point máximo de 4.02 e LOD escore multipoint 4.28 o que demonstra a presença de um novo lócus envolvido com o desenvolvimento da doença. O sequenciamento de nova geração de todos os éxons foi realizado nos nove indivíduos, presentes nas duas famílias, com gagueira desenvolvimental persistente familial. Análises de bioinformática permitiram filtrar, dentre diversas variações nucleotídicas, 20 variações presente em 19 genes, com potenciais implicações quanto ao fenótipo. Secundariamente, foram sequenciados, por sequenciador automático capilar, genes previamente apontados por outros estudos como envolvidos com a gagueira e outros distúrbios de fala e linguagem como o gene DRD2 (contendo as variações rs6275 e rs6277), GNPTAB, GNPTG, NAGPA, FOXP2 e CNTNAP2 em um grupo amostral de indivíduos com gagueira desenvolvimental persistente familial, não relacionados, e grupo controle. Diversas variações foram identificadas, porém sem consistência estatística significativa para afirmar que tais alterações são determinantes para a doença na população brasileira estuda. Em específico, não foram observados associação entre a variação rs6275 no gene DRD2 em casos de gagueira, publicados anteriormente.
ABSTRACT
Stuttering is a common disease that affects the fluency of speech, and is characterized by frequent repetitions or prolongations of sounds, syllables, or words, or by interruptions in the normal flow of speech. The disorder typically begins in children aged two to four years, and has an estimated incidence rate of around 5% of the population. Due to the high rate of spontaneous recovery, the estimated prevalence of the disorder is 1% in the general population. Despite the involvement of environmental factors, genetic factors have been shown to be critical to the development of the disease. Evidence supporting genetic factors include twin studies, adoption studies, family clusters of stuttering, and consanguineous relations in families with many cases of the disorder. However stuttering is a complex disorder in which the identification of the exact type of inheritance is difficult to determine because it does not follow Mendelian laws. The main goal of this study was to perform a genome-wide linkage analysis in 43 unrelated families from the Brazilian state of São Paulo, which had multiple cases of persistent developmental stuttering, in an effort to identify chromosomal regions containing possible causal genes, Linkage analysis was initially performed by genotyping of all families with chip-based methods that assayed 6056 sNP markers. Subsequent refinement of linkage locations used polymorphic microsatellite markers analyzed by capillary electrophoresis unsing an automated DNA sequencer. Unrelated normal Brazilian samples were used as a control group to estimate the allele frequencies of these markers in this population. Two families (BRPD_47 and BRPD_50) showed significant evidence for linkage in the region of chromosome 10q21 under a dominant inheritance model. Combining these two families produced a single point LOD score of 4.02 and a maximum multipoint LOD score 4.28. This demonstrates the presence of a new locus involved in stuttering development. Whole Exome Sequencing (WES) was performed in nine individuals present in these two families. Bioinformatics analysis enabled filtering of all observed variants to generate 20 variations in 19 genes as candidate causal variants. . Secondarily, we sequenced a number of genes that have previously been indicated as being involved in stuttering and other speech disorders, including DRD2 (containing variant sites rs6275 and rs6277),
GNPTAB, GNPTG, NAGPA, CNTNAP2 and FOXP2 in a sample of individuals with persistent developmental stuttering and controls. The sequencing of these genes identified was performed on an automated capillary sequencer. Several variations were identified, but no statistically significant evidence was found to assert that such changes are critical for the phenotype in the population studied. In particular, no support was obtained for the association of the rs6277 variant in the DRD2 gene with stuttering that had been previously published.
1. INTRODUÇÃO
1.1. DEFINIÇÃO DE GAGUEIRA
A gagueira é uma doença comum que afeta a fluência da fala e que se caracteriza
por repetições ou prolongamentos frequentes de sons, sílaba, palavras, ou por
hesitações, ou interrupções no fluxo normal da fala, bem como por: a) perturbação na
fluência e padrão de tempo normal da fala (inapropriado para a idade do indivíduo); b)
perturbação na fluência a qual interfere no rendimento escolar e profissional ou na
comunicação social; c) em presença de um déficit motor da fala, déficit sensorial, as
dificuldades na fala excedem aquelas habitualmente associadas com estes problemas
(ORGANIZATION, 2010).
A gagueira é um distúrbio complexo de comunicação oral, de caráter
multidimensional que muitas vezes se inicia na infância (SASSI;
CAMPANATTI-OSTIZ; ANDRADE, 2001; YAIRI, 1997) e que frequentemente é vivenciada pelo
indivíduo como uma perda de controle da fluência de sua própria fala (COOPER, E. B.,
1993; PERKINS, W. H.; KENT; CURLEE, 1991).
Fluência é um aspecto da produção da fala que se refere à continuidade,
suavidade, velocidade e/ou a força com que as unidades da linguagem (fonológico,
lexical, morfológico e/ou sintaxe) são expressas (GORDON W. BLOOD et al., 1999).
Segundo Brandi (1990), a fluência é caracterizada pela sequência (organização temporal
dos fonemas), pela duração (tempo necessário para a articulação do elemento fonético),
pela velocidade (rapidez com que os elementos fonéticos são articulados) e, pelo ritmo
(forma da velocidade da fala). Assim, alterações em qualquer um destes componentes
podem caracterizar as disfluências. O termo disfluência é tanto empregado para as
interrupções típicas ou comuns, quanto para as atípicas.
Todos os falantes apresentam momentos de disfluência na fala, frequentemente
caracterizada como típicas (BLOODSTEIN; RATNER, 2008; CORDES; INGHAM,
1995; DEGIOVANI; CHIARI; SCHAFFER, 1999; DUCHIN; MYSAK, 1987;
LEEPER; CULATTA, 1995; PERKINS, W.H., 1990; THRONEBURG; YAIRI;
PADEN, 1994; VAN RIPER; EMERICK, 1990; YAIRI; AMBROSE, 1992a; YAIRI;
AMBROSE; NIERMANN, 1993) as quais são tidas como incertezas linguísticas
relacionadas à formulação das frases ou a pronúncia das palavras (PERKINS, W.H.,
Entretanto, as disfluências atípicas estão relacionadas ao comportamento dos
indivíduos gagos (BLOODSTEIN; GROSSMAN, 1981; BLOODSTEIN; RATNER,
2008; DEGIOVANI et al., 1999; LEEPER; CULATTA, 1995; PETERS; GUITAR,
1991; RILEY, G. D., 1972; SCHWARTZ; CONTURE, 1988; THRONEBURG et al.,
1994; YAIRI; AMBROSE, 1992a; YAIRI et al., 1993). Certos comportamentos
podem definir a gagueira: repetição ao nível do fonema, da sílaba ou do sintagma;
alongamento de sons; bloqueios na fonação; posições articulatórias fixas; pausas
silenciosas; frases incompletas; inserção de sons estranhos à fala; mudanças súbitas na
tonalidade e na intensidade da voz; falha no ritmo; falta de sincronização entre a
respiração e a fonação; introdução sistemática de pequenas frases ou interjeições;
esforço motor durante a fala.
Além disso, pode-se observar também a presença de comportamentos associados,
chamados de “secundários”, “compensatórios” ou “acessórios” e que envolvem
comportamentos de evitação (não falar quando deseja fazê-lo, utilizar sinônimos para
palavras temidas, parafrasear a emissão pretendida), artifícios de atraso (utilizar
palavras sem significado ou palavras de preenchimento ou esperar para tentar falar),
iniciadores (piscar os olhos, inspirar fundo antes de iniciar a fala, antecipar um
momento de fala para iniciar a emissão da palavra temida), reações de disfarce (cobrir a
boca simulando uma tosse para esconder o fato de estar gaguejando), reações de
interrupção (sacudir a cabeça ou fazer caretas para sair de um bloqueio) e movimentos
de busca (usar hesitações ou, vogal inapropriada ou, alterar a velocidade de sons e
sílabas repetidas) (SASSI et al., 2001).
Apesar de não ter ainda suas causas bem compreendidas, muitos casos de gagueira
apresentam fortes evidências do envolvimento de fatores genéticos (ALM, 2006;
AMBROSE; YAIRI; COX, 1993; ANDREWS et al., 1991; FELSENFELD et al.,
2000; HOWIE, 1981; OOKI, 2005). Há indícios de que indivíduos com gagueira
recuperada, quando comparados a indivíduos com gagueira persistente podem ter, em
parte, bases genéticas distintas (DWORZYNSKI et al., 2007) motivando nos estudos
em que se levam em conta as atribuições genéticas, considerar qual o subtipo de
1.2. CLASSIFICAÇÃO DA GAGUEIRA
A gagueira pode ser classificada como desenvolvimental, neurogênica e
psicogênica; sendo a primeira a forma mais comum (PRASSE; KIKANO, 2008).
A gagueira desenvolvimental é inicialmente observada em crianças com idade
entre três e oito anos e, acomete mais de 80% da população em geral (LUDLOW,
2000). Por volta de 75% das crianças em idade pré-escolar, portadoras da gagueira
desenvolvimental, apresentam recuperação em quatro anos (YAIRI; AMBROSE, 1999).
Os pacientes com esse tipo de gagueira inicialmente apresentam sinais brandos que ou,
se recuperam ou, evoluem para um quadro clínico mais severo com o desenvolvimento
de comportamentos secundários (PRASSE; KIKANO, 2008).
As outras duas formas distintas de manifestação da gagueira são ou, por meio de
lesões cerebrais oriundas de eventos traumáticos, o que a classifica como neurogênica
ou, atribuída aos aspectos psicogênicos (GRANT et al., 1999) capazes de alterar o
padrão de fluência do indivíduo, geralmente adultos, a partir de um evento emocional
traumático (MAHR; LEITH, 1992).
Um dos grandes desafios até o momento tem sido diferenciar na infância, as
crianças que começam a gaguejar e que apresentam alto risco de se tornarem gagos
desenvolvimentais persistentes, daquelas propensas a se recuperarem espontaneamente
ao longo do tempo.
Pautados em estudos preliminares de caráter longitudinal para o
acompanhamento do desenvolvimento da fala em crianças, Yairi et al. (1996)
caracterizaram os sinais de gagueira desenvolvimental adotando os seguintes critérios:
a) idade inferior a seis anos; b) considerada pelos pais como gaga; c) considerada por
dois investigadores como gaga; d) tempo de duração da gagueira menor que 12 meses;
e) severidade da gagueira avaliada pelos pais com valores de pelo menos dois entre oito
pontos (0 = normal, 1 = limítrofe ;.... 6 = severo, 8 = muito severa); f) nível de
severidade avalido pelos pesquisadores, como maior ou igual a dois; g) exibir no
mínimo 3% de disfluências atípicas (ou gagas); h) não apresentar qualquer distúrbio
neurológico ou outras anomalias.
Desta forma, Yairi et al. (1996)puderam classificar os indivíduos gagos em três
distintos grupos:
1. Gagueira desenvolvimental persistente: presente por 36 meses ou mais após o
2. Gagueira desenvolvimental de recuperação tardia: presente, porém recuperada
entre 18 e 36 meses após o início do distúrbio;
3. Gagueira desenvolvimental de recuperação precoce: presente, porém
recuperada entre 18 meses ou menos após o início do distúrbio.
Figura 1. Classificação da gagueira, modificado de Oliveira et al (2012).
1.3. DIAGNÓSTICO DA GAGUEIRA
Avaliar a fluência da fala é essencial para o diagnóstico da gagueira e para isso
três aspectos devem ser considerados segundo Andrade et al. (2004):
1. Tipologia das rupturas
x Relacionadas com o processamento da linguagem ou da fala; 2. Velocidade de fala
x Fluxo de palavras por minuto - mede a taxa de produção de informação;
x Fluxo de sílabas por minuto - mede a taxa de velocidade articulatória;
3. Frequência de rupturas
x Avalia a porcentagem de descontinuidade da fala - mede a taxa de rupturas no discurso;
x Avalia a porcentagem de disfluências gagas - mede a taxa de rupturas
sugestivas de gagueira;
Gagueira
Neurogênica Psicogênica Desenvolvimental
Familial
Persistente
Recuperação tardia
Recuperação precoce
Isolada
Persistente
Recuperação tardia
Baseando-se nesses critérios, diversas abordagens podem ser utilizadas para o
diagnóstico dos distúrbios da fluência. Uma delas é a coleta de amostras de fala, por
meio de gravações de áudio e vídeo. Nesta metodologia avalia-se a fala auto-expressiva
obtida num mínimo de 200 sílabas expressas (fluentes), a partir de um estímulo visual
por figura (adaptada às diferentes faixas etárias). Assim, o diagnóstico de gagueira é
atribuído aos indivíduos que apresentarem a partir do Stuttering Severity Instrument for
Children and Adults – SSI-3 (RILEY, G.D, 1994) um escore referente ao nível de
classificação - gravidade leve (varia com a idade) e concomitantemente, apresentarem
no mínimo 3% de rupturas gagas (6 rupturas/200 sílabas expressas) (YAIRI;
2. REVISÃO DA LITERATURA
A gagueira é considerada uma doença herdável desde a década 1930 uma vez que
trabalhos daquela época já apontavam para a sua natureza familial (BERRY, 1938;
BRYNGELSON, 1939; BRYNGELSON; RUTHERFORD, 1937).
As informações quanto à epidemiologia e a herdabilidade da doença quando
combinados, através de métodos estatísticos de análise, com dados genéticos
(marcadores moleculares, regiões cromossômicas, genes) tem sido usados para o
entendimento das bases biológicas envolvidas.
2.1. INCIDÊNCIA E PREVALÊNCIA
A gagueira desenvolvimental comumente afeta pessoas de todas as etnias,
nacionalidades e classes sociais; surge na infância em crianças com idade entre dois e
quatro anos, apresentando uma taxa de incidência estimada em torno de 5% da
população (BLOODSTEIN; RATNER, 2008). Estudos tem mostrado também que essa
doença tem variado conforme o gênero dos indivíduos afetados. Estima-se que para o
sexo masculino seja ao redor de 4% enquanto que no feminino, 2% (ANDREWS et al.,
1983; ANDREWS; HARRIS, 1964; KIDD, K. K., 1978).
Entretanto, por ser uma doença que apresenta uma taxa de recuperação
espontânea entre 75 a 80% e, mais comum entre o sexo feminino, faz com que a
gagueira desenvolvimental persistente apresente uma prevalência estimada de 1% na
população (AMBROSE et al., 1993; BLOODSTEIN; RATNER, 2008; CRAIG, A. et
al., 2002; FELSENFELD, 2002; YAIRI; AMBROSE, 1999). Além disso, observam-se
também variações na estimativa das taxas de recuperação conforme a idade: pré-escolar
2,4%; escolar 1% e menor que 1% em adultos (BLOODSTEIN; RATNER, 2008).
Variações nas estimativas da prevalência observadas em estudos populacionais
(Tabela 1) são atribuídas a possíveis erros metodológicos ou de amostragem. Um ponto
crítico está relacionado, por exemplo, a idade dos indivíduos analisados, ao gênero e à
Tabela 1. Variações nas estimativas de prevalência em diferentes estudos e populações.
Estudo População Prevalência
estimada
(PORFERT; ROSENFIELD, 1978) Estudantes universitários americanos 2,1% (VAN RIPER, 1982) Crianças japonesas em idade escolar 0,8% a 1%
(ANDRADE, 1997) Crianças da cidade de São Paulo 2,9% (BLOODSTEIN, 1995; CRAIG, A. et al., 2002) População Huterita 2,2% (MANSSON, 2000) Ilha dinamarquesa de Bornholm 1% (MCKINNON; MCLEOD; REILLY, 2007) Estudantes de escola primária australiana 0,33%
(DWORZYNSKI et al., 2007) Gêmeos britânicos 1 a 3%
Uma das explicações possíveis, relacionada à prevalência maior de indivíduos
do sexo masculino com gagueira desenvolvimental persistente está baseada na teoria
hormonal proposta por Geschwind e Galaburda (1985). O hormônio sexual masculino,
testosterona, é responsável por promover um atraso no desenvolvimento neural do feto.
O hemisfério cerebral direto tende a se desenvolver naturalmente mais rápido em
relação ao hemisfério esquerdo, envolvido com a fala. Assim, o hemisfério cerebral
esquerdo de um feto do sexo masculino, estará exposto à ação de elevados níveis de
testosterona por mais tempo do que no feto feminino o que proporcionará a pricípio um
“excessivo” atraso no desenvolvimento do lado esquerdo afetando assim, mais o
desenvolvimento da fala em homens do que em mulheres.
Desta forma, pode-se estimar a razão sexual Masculino : Feminino de acordo
com as seguintes faixa etárias: 1M : 1F em crianças de dois a três anos (AMBROSE;
COX; YAIRI, 1997; YAIRI, 1983); 2,1M : 1F em crianças de dois a seis anos (YAIRI;
AMBROSE, 1992a) e de 4 a 5M : 1F entre os adolecêntes e adultos (BLOODSTEIN;
RATNER, 2008; BUCHEL; SOMMER, 2004; FELSENFELD, 2002; YAIRI;
AMBROSE, 1999).
2.2. ETIOLOGIA
A complexidade da gagueira, em diferentes instâncias, faz dela um problema
claramente, multidimensional e multicausal não permitindo ser tratada como uma
entidade nosológica única (SASSI et al., 2001)
A origem da gagueira, apesar de ainda não estar esclarecida, pode ser segundo
Brito Pereira (2002)o resultado da interação de três fatores predisponentes: emocional,
social e biológico. O emocional pode facilitar o estabelecimento da gagueira por meio,
deficiências na comunicação que gera ansiedade, incerteza e medo nos portadores de
gagueira, predispondo-os ao desenvolvimento de um autoconceito negativo.
Crianças com gagueira desenvolvimental em idade pré-escolar, apresentam mais
descontroles emocionais e problemas de atenção quando comparadas a crianças da
mesma idade, porém fluentes (KARRASS et al., 2006).
O vínculo entre a gagueira e os fatores biológicos predisponentes não está ainda
bem caracterizado, no entanto, são crescentes as evidências que sustentam a relação
entre a condição genética do indivíduo e a doença. Há mais de 70 anos a predisposição
genética para a gagueira é reforçada por referências à agregação familial da doença
(BRYNGELSON; RUTHERFORD, 1937; GRAY, 1940; MEYER, 1945; WEPMAN,
1939; WEST; NELSON; BERRY, 1939). Estudos analisando gêmeos monozigóticos
estimam que 70% das variações observadas em gagos são atribuídas a componentes
genéticos e o restante (30%) às influências ambientais (FELSENFELD et al., 2000).
Um amplo número de estudos de neuroimagem (Tabela 2) apontam para
diferentes regiões do sistema nervoso central, que uma vez alteradas, podem ser
responsáveis pelo desencadeamento da doença. Há um consenso de que em portadores
de gagueira desenvolvimental persistente, pequenas mudanças na arquitetura cerebral
devam ser atribuídas a fatores genéticos (FOUNDAS et al., 2001; YAIRI; AMBROSE,
Tabela 2. Estudos de neuroimagem e a identificação de possíveis regiões relacionadas à gagueira desenvolvimental persistente.
Estudos *Métodos Áreas nervosas
Sommer et al. (2002) (1)RM / (2)DTI Região cortical senso-motora.
Cykowski et al. (2008) RM Região perisilviana e assimetria cerebral.
Watkins et al .(2008) (3)RMf / RM / DTI Regiões motoras corticais e subcorticais (córtex ventral pré-motor).
Chang et al. (2008) RM / DTI / (7)VBM
Déficit de desenvolvimento da massa branca em torno das regiões motoras oro-faciais e redução da massa cinza na região
de Broca, ambos no hemisfério esquerdo.
De Nil et al. (2008) RMf
Baixa atividade do giro temporal esquerdo; aumento de atividades no giro temporal superior medial esquerdo, na insula
direita, córtex motor primário e córtex motor.
Chang et al. (2009) RMf Alterações na ativação de integração audio-motora.
Lu; Ning et al. (2009) RMf
Desconexão funcional do giro frontal inferior esquerdo para as áreas motoras esquerda, alteração na conectividade do circuito
basal cortical-ganglio-talâmico, alterações na integração cerebelar e diversas regiões fronto-parietais.
Lu; Peng et al. (2009) RM / DTI / VBM / (6) SEM
Alteração de conectividade no circuito basal gânglio-talamo-cortical.
Cykowski et al. (2010) RM / DTI Redução de massa branca na região perisilviana esquerda e, defasagem da mielinização em algumas regiões.
Choo et al. (2011) RM / DTI / VBM Anormalidades anatômicas no corpo caloso.
Chang et al. (2011) RMf Presença de diferenças anatômicas e funcionais entre a região frontal inferior esquerda e o córtex pré-motor.
Choo et al. (2012) RM / DTI / VBM Achados inconclusivos.
Jiang et al. (2012) RMf Alta atividade do córtex frontal inferior esquerdo e precuneus bilateral.
Ingham et al. (2012) RM / (5)PET Achados inconclusivos
O envolvimento de fatores genéticos com a gagueira desenvolvimental
persistente deve ser contabilizado no diagnóstico, prognóstico, tratamento e orientação
desses pacientes. O clínico pode ser alertado pelo paciente quando há familiares gagos e
como tal, uma história familiar é um bom indicador de um fator genético atuando no
desenvolvimento da doença.
2.3. FATORES GENÉTICOS
Os principais argumentos que fundamentam o envolvimento de fatores genéticos
com a gagueira desenvolvimental persistente são:
x A gagueira desenvolvimental persistente ocorre em famílias (AMBROSE et al.,
1997; AMBROSE et al., 1993; ANDREWS; HARRIS, 1964;
BRYNGELSON; RUTHERFORD, 1937; JOHNSON, 1959; KIDD, K., 1984;
KIDD, K. K., 1977;1978; KIDD, K. K.; REICH; KESSLER, 1973; PADEN;
YAIRI; AMBROSE, 1999; WATKINS, R. V.; YAIRI; AMBROSE, 1999);
x Estudos com gêmeos e envolvendo adoções - maior concordância entre gêmeos monozigóticos (62,5% a 90%) do que em dizigóticos (6,6% a 9%) (ANDREWS
et al., 1991; BLOODSTEIN; RATNER, 2008; DWORZYNSKI et al., 2007;
FELSENFELD et al., 2000; FELSENFELD; PLOMIN, 1997; HOWIE, 1981;
OOKI, 2005; RAUTAKOSKI et al., 2012);
x Relações de consanguinidade - a gagueira desenvolvimental persistente é mais
propensa a se desenvolver em indivíduos consanguíneos quando comparados a
casos em que os indivíduos afetados não tenham essa relação (ANDREWS et
al., 1991; FELSENFELD et al., 2000; FELSENFELD; PLOMIN, 1997);
Tais indicadores não só reforçam como também sustentam o direto envolvimento
dos componentes genéticos em diferentes populações, ao ponto de permitir estimar o
grau de contribuição. O fator herdabilidade (h2) mensura o grau de contribuição dos
fatores genético no desenvolvimento da doença. Alguns trabalhos (ANDREWS et al.,
1991; FAGNANI et al., 2011; FELSENFELD et al., 2000) apontam para valores de
herdabilidade estimados em 70%. Mais recentemente, Rautakoski et al. (2012) ao
analisarem 1728 gêmeos finlandeses gagos desenvolvimentais estimaram a
entre os gêmeos para o desenvolvimento da gagueira desenvolvimental foi de 82%
enquanto que os fatores ambientais, não compartilhados, porém envolvidos, 18%.
No entanto, a identificação de genes que contribuam para o desenvolvimento de
características altamente complexas, como a gagueira desenvolvimental persistente, tem
se tornado muito mais desafiadora do que os estudos genéticos de doenças com traços
mendelianos (BOTSTEIN; RISCH, 2003). Diversos fatores estão envolvidos neste
problema como a heterogeneidade etiológica e genética e, portanto a necessidade do uso
de modelos genéticos complexos, com muitas variáveis para efeitos de lócus, interações
gene-gene e gene-ambiente de forma a justificar o uso de diversos métodos e modelos
estatísticos para as análises. O método adequado de análise genética para a gagueira
desenvolvimental persistente familial exige uma combinação de passos para a
identificação de regiões cromossômicas, contendo genes possívelmente responsáveis
por contribuir com o entendimento da complexa etiologia da doença
(WITTKE-THOMPSON et al., 2007).
O exato modelo de transmissão da herança para a gagueira desenvolvimental
persistente familial, até então, não está claramente definido, além de poder ser diferente
entre populações distintas. Há trabalhos que sugerem a existência de um gene principal
responsável por aumentar o risco de ocorrência da gagueira uma vez combinado com
outros genes (YAIRI; AMBROSE, 2005).
Ao estudar famílias com gagueira desenvolvimental persistente, Viswanath et al.
(2004) sugeriram um modelo de transmissão autossômica dominante sob influência de
um gene principal combinado por duas variáveis: o gênero e a condição fenotípica dos
pais. Nestas condições, em indivíduos gagos, porém sem os pais afetados, a penetrância
estimada foi de 37% em homens e, 7% nas mulheres. No entanto, nos casos em que os
pais eram também afetados as estimativas aumentaram para 67% nos homens e, 19%
nas mulheres.
Diferentes famílias paquistanesas consanguíneas analisadas em situações
distintas, também apresentaram indefinições quanto ao exato modo de herança. Na
família paquistanesa PKST77, Raza et al. (2010) verificaram um modelo de herança
autossômica recessiva com penetrância completa, porém posteriormente na família
PKST58, Raza et al. (2011) observaram o mesmo modelo de herança, no entanto com
2.4. DOENÇAS COMPLEXAS
O termo doenças complexas é atribuído a uma condição em que o conjunto de
traços fenotípicos resulta da combinação de componentes genéticos, ambientais e
sociais. Trata-se de uma condição genética na qual a identificação exata do tipo de
herança é difícil de ser determinada uma vez que não seguem as leis de Mendel. Alguns
fenômenos biológicos colaboram para dificultar o entendimento dos mecanismos
envolvidos nesta condição como variações na penetrância e na expressividade gênica
bem como, indefinição do próprio fenótipo, traços poligênicos, interações gene-gene e
gene-ambiente e que uma vez combinados a casos de agregação familiar reforçam o
envolvimento genético (CRAIG, J., 2008; HAINES; PERICAK-VANCE, 1998).
2.4.1. Estratégias de análise
Diferentes estratégias de análise genética tanto para as doenças mendelianas
quanto para as de ordem complexa têm sido usadas por pesquisadores para identificar e
entender os fatores genéticos envolvidos no desenvolvimento e transmissão dessas
doenças.
A clássica estratégia por clonagem posicional, adotada para muitas doenças
mendelianas, consiste na identificação e clonagem de um gene baseando-se unicamente
na sua posição no genoma. Usualmente, tal abordagem envolve uma sequência de
passos como a identificação de famílias afetadas, coleta de amostras biológicas para
purificação do DNA, genotipagem de marcadores moleculares polimórficos, análises de
ligação, localização da região cromossômica candidata, refinamento do mapa genético,
obtenção do mapa físico e por fim, a identificação do gene (HAINES;
PERICAK-VANCE, 1998).
No entanto, quando a estratégia de análise envolve o mapeamento de doenças com
características complexas, como a gagueira desenvolvimental persistente familial, a
sequência linear adotada pela clássica abordagem, pode sofrer sinuosas modificações. A
identificação de genes neste contexto é trabalhosa, uma vez que vários genes podem
estar envolvidos com o fenótipo além de os fatores ambientais serem de difícil controle.
Uma vez desconhecida a patogênese sobre a qual diversos genes podem estar
envolvidos, a realização de uma varredura sistemática do genoma para a detecção de
ligação pode ser uma estratégia inícial (CORREIA, 2008). No entanto, a escolha do
características específicas e, portanto, da caracterização dos indivíduos analisados.
Assim, falhas na caracterização adequada dos pacientes portadores da doença, nesta fase
do estudo, podem implicar em exclusões ou mesmo substituições na amostra que
interferem diretamente na dinâmica das análises e dos resultados (HAINES;
PERICAK-VANCE, 1998).
A figura 2 representa esquematicamente as etapas que podem ser seguidas para o
estudo tanto de doenças comuns quanto nos casos de ordem complexa. Cada uma dessas
fases apresentam peculiaridades e fatores que devem ser levados ou não em
consideração, principalmente nas doenças complexas as quais a melhor forma de
abordagem envolve a análise criteriosa de traços específicos (HAINES;
Definição do fenótipo *Consistência
*Rigor
Identificação de evidências do envolvimento de fatores genético *Estudos com gêmeos
*Risco relativo *Análise de segregação *Estudos envolvendo adoções *Herdabilidade
Definição da estrutura do experimento a partir de uma ou mais formas de abordagem:
Pares de irmão Grandes famílias Único membro afetado na família
Amostragem, armazenamento das amostras, coleta de dados *Histórico da família
*Informações clínicas *Coleta do DNA
Genotipagem
Análise dos dados
Análise paramétrica: análise de LOD escore
Análise não paramétrica: análise de par de
irmãos; análise de parentes
afetados
Estudos de associação Caso controle baseado
em famílias
Identificar, testar e mapear regiões de interesse
Mapeamento físico Identificação do gene
Definição das interações *gene-gene
*gene-ambiente
1
2
3/4
4a 4b 4c
5
6
7
7a 7b 7c
8/9/10
11/12
13
A análise de ligação é um importante instrumento no mapeamento de doenças
complexas e, baseia-se em verificar, dentro de uma família, a cosegregação de
marcadores genéticos e o fenótipo da doença e, analisar se ambos estão fisicamente
ligados. Há ligação quando existe uma proximidade mensurável entre os marcadores
envolvidos, ao longo de um mesmo cromossomo, e a doença. As análises de ligação
recebem apoio estatístico na medida em que as estratégias de estudo são estabelecidas.
Geralmente, nos casos de doenças humanas envolvendo famílias, é frequente a falta de
algumas informações, sendo necessário então estimar a ligação a partir de dados
familiais incompletos (FEITOSA; KRIEGER, 2002).
A medida direta da ligação genética é obtida pela fração de recombinação, a qual
analisa a capacidade dos pais produzirem descendentes recombinantes devido à
permutação dos cromossomos homólogos durante a meiose. A fração de recombinação
ș (teta) é designada como sendo a proporção de meioses em que ocorre a recombinação e indica a distância genética entre dois loci (BURTON; TOBIN; HOPPER, 2005). Os
valores de ș variam de 0 (zero) para loci que estão completamente ligados a 0,5, para loci não ligados ou no mesmo cromossomo ou, em cromossomos distintos (segregação
independente) (HAINES; PERICAK-VANCE, 1998).
Supondo-se que numa família afetada observemos a segregação consistente de um
marcador genético com a doença, pode-se entender que há um lócus de susceptibilidade
para a doença na região desse marcador (DAWN TEARE; BARRETT, 2005).O cálculo
da fração de recombinação (ș) é feito a partir da razão entre o número de recombinações (k) e o número total de eventos numa meiose (n), recombinantes e não recombinantes,
assim ș = k/n (PASTERNAK, 2005).
No entanto, entre grandes distâncias, diversas permutas podem acontecer o que
diminui a eficiência do cálculo da fração de recombinação. As funções de mapeamento
são uma alternativa para auxiliar nos cálculos do valor real de ș, sendo que duas merecem destaque: a função de Haldane (1919) e a função de Kosambi (1944). A
primeira assume que não há interferência, ou seja, a permutação acontece
uniformemente ao longo de todo o cromossomo enquanto que, a segunda leva o
fenômeno de interferência em consideração nos cálculos e por isso é a função mais
frequentemente usada nos mapeamentos do genoma humano (HAINES;
PERICAK-VANCE, 1998). A interferência é o fenômeno pelo qual a ocorrência de permuta em
uma região do cromossomo é capaz de interferir reduzindo a taxa de permuta na região
As análises de ligação podem ser de dois tipos: paramétrica no qual o modelo de
herança é pré-determinado ou, não paramétrica, livre de modelo.
2.4.1.1. Análise de ligação paramétrica
As análises baseadas num modelo, a probabilidade de ligação é estimada pelo valor
de LOD (logarithm of odds ratio) escore, atribuído ao logaritmo na base 10 de uma
razão de verossimilhança, desenvolvido por Morton (1955)como método estatístico.
A razão de verossimilhança é a probabilidade de se observar numa família a
distribuição de genótipos assumindo a existência de ligação versus a mesma
probabilidade estimada, porém com segregação independente (ș = 0.5). Assim, quanto maior o valor de LOD escore, maior será a evidência de ligação entre o marcador e o
fenótipo analisado para características mendelianas. O LOD escore maior ou igual a 3, é
considerado como evidência significativa de ligação, uma vez que a hipótese alternativa
testada tem uma verossimilhança mil (103) vezes maior que a hipótese nula com ș = 0,5. Nesse sentido, valores de LOD escore entre 1,5 e menores que 3 sugerem a existência
de ligação, enquanto que valores iguais ou menores que -2 indicam a inexistência de
ligação (FEITOSA; KRIEGER, 2002) podendo ser usados como mapeamento de
exclusão.
A partir desse princípio, Elston & Stewart (1971) elaboraram um algoritmo capaz
de calcular a razão de verossimilhança em grandes genealogias com diversas gerações,
porém limitado quanto ao número de marcadores genéticos analisados simultaneamente.
Para aumentar a velocidade das análises envolvendo diversos alelos em apenas dois loci
para análises de genealogias complexas, Ott (1974) a partir da mesma base algoritma
desenvolveu programa de computador (LIPED) para estimar a razão de verossimilhança
(LOD escores).
Posteriormente, outros programas foram desenvolvidos utilizando ou, a mesma
base algoritma, como Linkage (LALOUEL; LATHROP; WHITE, 1986) e o FastLink
(COTTINGHAM; IDURY; SCHAFFER, 1993; SCHAFFER et al., 1994) ou, diferentes
algoritmos, como de Lander & Green (1987) que permitiu analisar genealogias
2.4.1.2. Análise de ligação não paramétrica
O método de análise livre de modelo, não requer, a princípio, parâmetros que
determinem o tipo de herança da doença analisada muito embora seja fundamental a alta
“qualidade” na definição e caracterização do fenótipo da doença (HAINES;
PERICAK-VANCE, 1998). Neste modelo, fatores como frequências gênicas e penetrância, não
precisam ser estabelecidas num primeiro instante uma vez que funções dessas variáveis,
bem como da taxa de recombinação (ș) podem ser estimadas, concomitantemente (FEITOSA; KRIEGER, 2002).
A análise de ligação não paramétrica é medida a partir do cálculo de
compartilhamento dos alelos de um marcador conhecido e a doença (HAINES;
PERICAK-VANCE, 1998). Um dos métodos mais conhecidos é o método de pares de
irmãos (ELSTON, 2000; HASEMAN; ELSTON, 1972; PENROSE, 1935; RISCH,
1990) que se baseia no conceito de identidade por descendência (IBD - Identity by
Descent) quantificando a semelhança genética, entre pares de indivíduos. As relações de
parentesco, como nos pares de irmãos, podem compartilhar zero, um ou dois alelos
oriundos de um ancestral em comum (FEITOSA; KRIEGER, 2002). Assim, o teste visa
basicamente comparar as proporções esperadas e observadas para esses alelos e então,
identificar aqueles marcadores compartilhados por descendência, mais frequentemente
que o esperado, entre os pares de irmãos.
Embora o método de análise não paramétrica seja menos robusto, quando
comparado ao paramétrico, no processo de identificação de ligação de marcadores
genéticos e o fenótipo, situações envolvendo doenças de ordem complexa na qual pouco
se sabe a respeito, a adoção desse método, num primeiro momento, é uma estratégia a
ser considerada (HAINES; PERICAK-VANCE, 1998).
2.4.1.3. Estudos de associação
Os estudos de associação também buscam identificar a contribuição genética
envolvida num fenótipo com base numa determinada população. Frequentemente,
envolvem a comparação das frequências alélicas do lócus marcador entre o grupo
afetado e o grupo controle. Uma vez observadas diferenças significativas entre as
frequências pode-se dizer que há associação entre a doença e o(s) alelo(s) (HAINES;
O cálculo das frequências alélicas é feito a partir da genotipagem de marcadores
densamente espaçados ao longo do genoma tendo em vista a captura de variações
comuns à população. Assim, partindo-se deste princípio e utilizando um grande
conjunto de amostras (grupo afetado e grupo controle) é possível mapear variações no
genoma, comuns à população estudada e de efeito modesto, susceptíveis à doença
(FEITOSA; KRIEGER, 2002).
Entretanto, um estudo mal planejado, envolvendo análise de associação pode gerar
problemas como, por exemplo, ao se estudar uma população estratificada por etnias. A
estratificação pode levar a resultados de associação equivocada do lócus “associado” à
doença uma vez que há desde suas origens uma associação intrínseca do mesmo, própria
da população. Uma solução para isso pode ser o uso de familiares não afetados pela
doença, como grupo controle (FEITOSA; KRIEGER, 2002).
Nas análises de associação é esperado que o grupo amostral e o controle se
diferenciem tanto pelo fenótipo quanto pela constituição genética, o que em grupos
etnicamente estratificados, um alelo específico e característico desta etnia pode
erroneamente ser identificado como fator de risco para a doença.
2.5. ANÁLISES DE LIGAÇÃO E A GAGUEIRA
Estudos envolvendo a análise de ligação têm sido aplicados na tentativa de
identificar genes envolvidos com a gagueira desenvolvimental persistente familial. No
entanto, por ser uma doença de caráter complexo, muito diferente das doenças tidas
como mendelianas, esse tipo abordagem é desafiada a cada trabalho, pois há fatores que
interferem quanto a eficiência desse método para a identificação clara dos componentes
genéticos envolvidos.
Até então há sete estudos (RAZA, M. H. et al., 2011; RAZA, M. HASHIM et
al., 2012; RAZA, M. H. et al., 2010; RIAZ et al., 2005; SHUGART et al., 2004;
SURESH et al., 2006; WITTKE-THOMPSON et al., 2007) descritos envolvendo a
gagueira desenvolvimental persistente familial e as análises de ligação.
A partir de 68 famílias de gagos (188 gagos e 38 fluentes = 226 indivíduos)
norte americanos ascendentes de europeus, Shugart et al. (2004) realizaram o primeiro
estudo de ligação com a gagueira desenvolvimental persistente familial. Os resultados
por eles apresentados sugeriram forte evidência de ligação (escore de 5,143), a partir de
disso, apesar de baixo, sugestivos NPL escores para os cromossomos 1, 13 e 16 foram
apontados.
Em 56 famílias paquistanesas consanguíneas (144 gagos e 55 fluentes = 199
indivíduos), Riaz et al. (2005) encontraram evidência de ligação com análises NPL nos
cromossomos 1, 5, 7 e 12. Com o refinamento do mapa de ligação a partir de
marcadores microssatélites a evidência de ligação no cromossomo 12 foi mantida, pois
tanto para a análise não paramétrica quanto paramétrica os escores foram de 4,61 e 3,51,
respectivamente. Os demais cromossomos apresentaram escores superiores a 2
sugerindo também possíveis ligações no entanto, a forte evidência encontrada em 12q
foi sugerida como a responsável por causar a doença que posteriormente foi confirmada
por Kang et al. (2010) a partir da identificação dos genes.
Em um terceiro estudo, Suresh et al. (2006) realizaram análise de ligação num
conjunto de 100 famílias de norte americanos de ascendência européia, suecos e
israelenses. A classificação das amostras foi feita de acordo com o fenótipo e separadas
em três grupos: gagueira desenvolvimental persistente – 252 indivíduos; gagueira
desenvolvimental recuperada – 45 indivíduos; muito jovens para o estabelecimento do
diagnóstico – 19 indivíduos. A análise de ligação foi aplicada e combinada em
diferentes etapas. Inicialmente as análises foram aplicadas num grupo (297 indivíduos)
envolvendo gagos desenvolvimentais persistentes e recuperados, obtendo-se evidência
sugestiva de ligação no cromossomo 9 (LOD escore 2,3), porém ao analisar apenas os
252 indivíduos com gagueira desenvolvimental persistente, o cromossomo 15 foi
indicado como sugestivo (LOD escore 1,95). Por fim, segregando as amostras de gagos
por gênero, a análise de ligação apresentou resultados mais expressivos: para gagos do
sexo masculino, o cromossomo 7 (LOD escore 2,99) e para gagos do sexo feminino, o
cromossomo 21 (LOD escore 4,5).
Ainda no mesmo trabalho, Suresh et al. (2006)ao observarem que a cada tipo de
abordagem analítica feita, diferentes regiões cromossômicas eram sugeridas e,
baseando-se em sugestões de que a gagueira desenvolvimental persistente poderia ser o
resultado da combinação de múltiplos genes (poligênica) foram aplicadas análises de
ligação condicional, com o objetivo de testar as áreas de ligação com o restante do
genoma.
Nas análises condicionais, as interações entre dois lócus podem ser tanto
positivas quanto negativas, quando o LOD escore condicional para um marcador sofre
cromossomos 2 e 9. No entanto, quando há um decréscimo do LOD escore condicional,
diz-se que há uma interação negativa, o que foi observado entre os cromossomos 7 e 12.
A partir de um isolado populacional com gagueira desenvolvimental chamado
Hutterites cuja origem envolveu um processo evolutivo conhecido como efeito
fundador, Wittke-Thompson et al. (2007) realizaram um novo estudo.
Baseados num diagnóstico sobre na impressão pessoal de cada um dos
envolvidos em relação à doença 48 indivíduos foram diagnosticados como gagos
desenvolvimentais, dos quais 12, recuperados e 36 persistentes. As primeiras análises de
ligação do genoma (GWL, genome-wide linkage) sugeriram evidências no cromossomo
3 (p value 0,013); 13 (p value 0,012) e 15 (p value 0,02). Novas análises, agora
envolvendo estudo de associação, apontaram para regiões sobrepostas às anteriormente
ligadas, além disso, para uma nova região. O cromossomo 3 (p value 0,0047), o 13 (p
value 0,0055) e o 9 (p value 0,0067).
A partir dos dados de Suresh et al. (2006), Wittke-Thompson et al. (2007)
realizaram um estudo de meta-análise em 105 famílias o que sugeriu dois novos
cromossomos como candidatos: o cromossomo 2 (p value 0,013) e o 5 (p values 0,0051
e 0,015).
Mais recentemente, três novos trabalhos integraram a lista de estudos de ligação
envolvendo a gagueira desenvolvimental persistente familial. A partir de uma família
paquistanesa consanguínea (PKST77) com gagueira desenvolvimental persistente, Raza
et al. (2010) identificaram ligação na região 3q13.31 (LOD escore 3,23). O elevado grau
de consanguinidade na população do Paquistão permitiu que o modelo de herança
autossômica recessiva fosse assumido nas análises paramétricas. A forte evidência de
ligação no cromossomo 3 foi confirmada a partir da genotipagem de marcadores
microssatélites e SNPs (Single Nucleotide Polymorphism) seguidos de nova análise. A
região 3q13.2-3q13.33 foi confirmada como ligada com a obtenção de um LOD escore
4,23 para a análise de ligação de ponto e, de 4,92 para análises multiponto.
Posteriomente, uma nova família paquistanesa consanguínea com gagueira
desenvolvimental persistente (PKST58) foi identificada com ligação para o fenótipo por
Raza et al. (2011) na qual dos 26 membros genotipados, 14 foram diagnósticados com
gagueira desenvolvimental persistente. A análise de ligação paramétrica sugeriu
ligações nos cromossomos 6, 9, 11, 16 e 21 com LOD escores variando de 2,1 a 2,76. O
refinamento do mapa de ligação (genotipagem de marcadores microssatélites) em cada
recessivo, confirmaram apenas a região 16q como candidata. Seguindo essas evidências
e aplicando-se análises de ligação multiponto, com o mesmo padrão de herança
recessivo modificado, um LOD escore de 4,42 foi obtido na região cromossômica
16q12.1-16q23.1.
O último estudo realizado até o momento é o de Raza et al. (2012). Neste
trabalho, a partir de uma família africana, proveniente da República de Camarões,
denominada CAMST01, foi realizado um extenso e complexo trabalho de mapeamento
genético envolvendo análises de ligação. Por tratar-se de uma grande família, rara e sem
evidências de consanguinidade, contendo 71 indivíduos, dos quais pelos menos 33
foram diagnosticados como gagos desenvolvimentais persistentes, foram aplicados, a
partir da genotipagem de marcadores SNPs e microssatélites, diferentes estratégias para
as análises de ligação. O heredograma da CAMST01 foi subdivido em cinco outros
menores, que permitiram além de facilitar as análises, em vista de limitações impostas
pelos programas, realizar combinações sistemáticas com as “subfamílias” facilitando a
identificação das regiões ligadas. Preliminarmente, as regiões 3q, 15q, 2p, 3p, 14q e 15q
apresentaram evidências de ligação. A partir do uso combinado das “subfamílias” e de
modelos de herança, como entre as regiões 2p e 15q, as análises estimaram um LOD
escore de 6,57. Além disso, diversos outros pontos do genoma apontaram para fortes
evidências de ligação (2,0 <. LOD escore < 3,0). Essa ampla distribuição de regiões ao
longo do genoma, possivelmente envolvidas, deve-se a presença de múltiplos alelos
com elevada penetrância em diferentes locis, uma vez que são observados, atipicamente,
grande número de afetados.
Um resumo de todos os estudos de ligação é apresentado na tabela 3. De todos
os cromossomos apontados por estes trabalhos, apenas nove diferentes cromossomos
apresentaram a sobreposição de evidências de ligação em pelo menos dois estudos. No
entanto, apenas um cromossomo (cromossomo 3) é tido com condições significativas de
ligação (RAZA, M. H. et al., 2010; WITTKE-THOMPSON et al., 2007). Até o
momento, de todos os trabalhos, apenas Riaz et al. (2005) foi capaz de servir como
referência e, auxiliar na descoberta de genes diretamente envolvidos com o
Tabela 3. Estudos de ligação desenvolvidos em famílias portadoras de gagueira desenvolvimental persistente. Trabalhos População Cromossomos com significativa evidência de ligação Outros cromossomos com evidências de
ligação
Shugart et al. (2004) Norte-americanos e
descendentes europeus 18 1, 2, 10, 13
Riaz et al. (2005) Paquistaneses
consanguíneos 12 1, 5, 7
Suresh et al. (2006)
Norte-americanos com ascendência europeia,
suecos e israelenses
9, 15, 2 (condicionalmente)
7 masculino sexo-específico e 21 feminino
sexo-específico
Wittke-Thompson et al. (2007)
Isolado populacional -
Hutterites 3, 13 2, 5, 9, 15
Raza et al. (2010) Família paquistanesa
consanguínea 3 -
Raza et al. (2011) Família paquistanesa
consanguínea 16 6, 9, 11, 21
Raza et al. (2012) Família africana
camaronesa 2, 15 3, 14
2.6. ESTUDO DE GENES CANDIDATOS
O mapeamento genético, por análise de ligação, tem por objetivo identificar
regiões em que estão presentes genes, tidos como candidatos, possivelmente envolvidos
com o fenótipo da doença. No entanto, outra forma de identificação de um gene é
através do entendimento prévio dos mecanismos biológico da doença sob investigação.
Conhecendo-se, por exemplo, informações bioquímicas como os tipos de enzimas
envolvidas num processo desencadeador da doença pode-se chegar a informações
quanto aos possíveis genes envolvidos e, portanto candidatos.
Para a realização do estudo de genes candidatos, geralmente utilizam-se dois
grupos de indivíduos: os afetados pela doença (grupo amostral) e os sem a doença
(grupo controle), o que configura o clássico estudo de caso-controle (KWON; GOATE,
2000).
O estudo de genes candidatos a partir da análise de casos e controles apresenta
algumas vantagens frente ao mapeamento por ligação. Primeiro, maior facilidade de