Universidade de Lisboa
Faculdade de Farmácia
An update on anticancer triazene compounds
Ana Rita Custódio Santos
Mestrado Integrado em Ciências Farmacêuticas
Universidade de Lisboa
Faculdade de Farmácia
An update on anticancer triazene compounds
Ana Rita Custódio Santos
Monografia de Mestrado Integrado em Ciências Farmacêuticas apresentada à Universidade de Lisboa através da Faculdade de Farmácia
Orientador: Doutora Ana Paula Francisco, Professora
auxiliar
Abstract
Triazenes are a very useful and diverse class of compounds that have been studied, principally, for their potential in the treatment of many tumors including brain, leukemia and melanoma. Novel compounds of this class continue appearing in the literature frequently as either anticancer compounds or even with other therapeutic interest. This monography focused on several types of triazenes from the simplest ones like 1,3-dialkyl-3-acyltriazenes to the more complex like combi-triazenes with emphasis on the mechanisms of their antitumor action and how can they be developed as effective antitumor agents. Although not all existing triazenes are addressed, those chosen largely represent the class and the latest discoveries.
While aliphatic triazenes are very sensitive to proteolytic decomposition and all of investigations are old, other triazenes reported have more clinical interest with good results in recent investigations. In the case of 1,3-diaryltriazenes, two types of molecules deserve special attention: alkyne analogues of diminazene aceturate (DMZ) and 4-nitro-substituted 1,3-diaryltriazenes. Aryl morpholino triazenes are other class of compounds recently investigated that could inhibit CYP1A1 and CYP1B1 at the micromolar level and could also have similar therapeutic value as resveratrol in preventing cancer. In the class of triazenoheterocycles, just one compound, was lately studied and demonstrated biochemical selectivity for EGFR and HER-2 receptors and significant reduction of cell growth in four human pancreatic cancer cell lines. Lastly, in relation to combi-molecules and specifically to combi-triazenes, a quite number of these molecules with dual targeting properties were developed and demonstrated anticancer activities both in vitro an in vivo, in the recent two decades. The most recent combi-triazene was designed to have a poly(ADP-ribose) polymerase (PARP) inhibitor.
Keywords: Triazene; Anticancer activity; 1,3-diaryltriazenes; Aryl morpholino
Resumo
Os triazenos são uma classe de compostos muito útil e diversificada e têm vindo a ser estudados, principalmente, devido ao seu potencial terapêutico em muitos tumores incluindo do cérebro, leucemias e melanoma. Frequentemente, novos compostos pertencentes a esta classe aparecem na literatura quer com a sua típica atividade anticancerígena ou mesmo com outros interesses terapêuticos. Esta monografia contém uma revisão destes compostos desde os mais simples como os 1,3-dialquil-3-aciltriazenos aos mais complexos como os combi-triazenos enfatizando os seus mecanismos de ação e o seu desenvolvimento como agentes antitumorais efetivos. Apesar de nem todos os tipos de triazenos existentes estarem contemplados nesta monografia, os escolhidos representam amplamente a classe e as novas descobertas. Enquanto que os triazenos alifáticos, pela sua estrutura, são muito sensíveis à decomposição proteolítica e todas as investigações já são antigas, outros triazenos possuem maior interesse clínico com bons resultados em estudos recentes. No caso dos 1,3-diariltriazenos duas moléculas merecem especial atenção: os análogos alcino do aceturato de diminazeno (DMZ) e os 1,3-diariltriazenos substituídos em para com um grupo nitro. Os aril morfolino triazenos são outra classe de compostos recentemente investigada que consegue inibir os CYP1A1 e CYP1B1 apresentando um valor terapêutico semelhante ao resveratrol na prevenção do cancro. Dentro dos triazenos heterocíclicos, apenas um composto foi estudado ultimamente, demonstrando seletividade bioquímica para os recetores EGFR e HER-2 e redução significativa do crescimento celular em quatro tipos de células de cancro do pâncreas. Por fim, várias moléculas quiméricas e especificamente os combi-triazenos, foram desenvolvidos nas últimas duas décadas sendo que o mais recente foi delineado para conter um inibidor da poli(ADP-ribose)polimerase (PARP).
Palavras-chave: Triazeno; Atividade anticancerígena; 1,3-diaryltriazenos; Aril
Acknowlegments
Tanto a elaboração desta monografia como o longo percurso de 5 anos só foram possíveis graças a um conjunto admirável de pessoas.
Um especial agradecimento à professora Doutora Ana Paula Francisco, orientadora desta monografia, pelo sorriso e amabilidade, pela transmissão de conhecimentos científicos e pela permanente disponibilidade.
Aos meus pais, avó e irmão pelos valores que sempre me transmitiram e pelo apoio incondicional em todas as fases do curso.
Ao meu namorado, André, que desde o primeiro dia me motivou a fazer mais e melhor com as suas sábias e amáveis palavras.
Às minhas 7 companheiras desta longa jornada pela amizade, pelos sorrisos e pela interajuda.
Abbreviations
ADP Adenosine diphosphate
AGT O6-alkylguanine-DNA alkyltransferase
AIC 5-aminoimidazole-4-carboxamide
ATP Adenosine triphosphate
BaP Benzo[a]pyrene
CBzM 1-(2-chloroethyl)-3-benzyl-3-(methylcarbamoyl)triazene
CMC 1-(2-chloroethyl)-3-methyl-3-carbethoxytriazene
CML Chronic myeloid leukemia
CMM 1-(2-chloroethyl)-3-methyl-3-(methylcarbamoyl)triazene
CR Cascade release
CYP Cytochrome P450
CYP1A1 Cytochrome P450 1A1
CYP1A2 Cytochrome P450 1A2
CYP1B1 Cytochrome P450 1B1
DMA 3-acetyl-1,3-dimethyltriazene
DMC 3-carbethoxy-1,3-dimethyltriazene
DMM 3-(N-methylcarbamoyl)-1,3-dimethyltriazene
DMZ Diminazene aceturate
DNA Deoxyribonucleic acid
dNu Deoxynucleotide
DTIC Dacarbazine
EGFR Epidermal growth factor receptor
EH Epoxide hydrolase
EMEA European Medicines Agency
FDA US Food and Drug Administration
HER-2 Human epidermal growth factor receptor 2
HMTIC 5-(3-hydroxymethyl-3-methyl-1-triazeno)imidazole-4-carboxamide
IC50 Half-maximal inhibitory concentration
M Molar concentration
MAP Mitogen-activated protein
MGMT O6-methylguanine-DNA methyltransferase
MRSA Methicillin resistant Staphylococcus aureus
MTIC 5-(3-methyl-1-triazeno)imidazole-4-carboxamide
nM nanomolar
O6-BG O6-benzylguanine
PARP poly(ADP-ribose) polymerase
PAHs Polycyclic aromatic hydrocarbons
Ph Philadelphia
RNA Ribonucleic acid
ROS Reactive oxygen species
SAPK/JNK Stress-activated protein kinase/c-Jun NH2-terminal kinase
TB Tuberculosis
TK Tirosine kinase
TMZ Temozolomide
Index:
1 Introduction ... 11
1.1 Triazenes in anticancer chemotherapy ... 11
1.2 Dacarbazine ... 12
1.3 Temozolomide ... 13
1.4 Goals of monography ... 15
2 Materials and methods ... 16
3 Prodrugs of aliphatic triazenes ... 17
3.1 1,3-dialkyl-3-acyltriazenes ... 17 3.1.1 Synthesis ... 18 3.1.2 Mechanism of action ... 20 4 1,3 – diaryltriazenes ... 23 4.1 Synthesis ... 23 4.2 Mechanism of action ... 24
4.2.1 Diminazene aceturate (Berenil) and derivatives ... 24
4.2.2 4-nitro-substituted 1,3-diaryltriazenes ... 27
4.3 Other therapeutic interests ... 28
4.3.1 Anti-mycobacterial activity ... 28
4.3.2 Antibacterial activity against MRSA ... 28
5 Aryl morpholino triazenes ... 30
6 Triazenoheterocycles ... 33
6.1 Synthesis ... 33
6.2 Properties ... 33
6.3 2-Triazenoazaindoles and pancreatic cancer ... 34
7 “Combi-triazenes” ... 36
7.1 Development of resistance and concept of hybrid drugs ... 36
7.2 EGFR TK inhibitor-linked DNA damaging agents ... 37
7.2.1 First generation of combi-molecules ... 37
7.2.2 Combi-molecules with improved properties ... 39
7.2.2.1 ZRBA1 as a radiosensitizer ... 40
7.2.2.2 JDE52: a bistriazene combi-molecule ... 40
7.2.3 Cascade-release targeting combi-molecules ... 41
7.2.4 Combi-molecule containing N-acetoxymethyl carbamate ... 43
7.2.5 Synthesis of EGFR TK inhibitor-linked DNA damaging agents ... 43
7.3 Bcr-Abl TK inhibitor-linked DNA-damaging agents ... 45
7.4 AGT inhibitor-linked DNA-damaging agents ... 49
7.5 PARP inhibitor-linked DNA damaging agents ... 50
8 Conclusion ... 53
Figure Index:
Figure 1 - Triazene general structure. ... 11
Figure 2 - General synthetic routes to triazenes. (3) ... 12
Figure 3 - Structure of dacarbazine. ... 12
Figure 4 - Structure of temozolomide. ... 13
Figure 5 - Mechanism of action of DTIC and TMZ. (7) ... 14
Figure 6 - General structure of 1,3-dialkyl-3-acyltriazenes. ... 17
Figure 7 - Synthesis of 1-(2-Chloroethyl)-3-methyl-3-acyltriazenes. (22,23) ... 19
Figure 8 - Decomposition of 1-(2-chloroethyl)-3-acyl-3-methyltriazenes. (24) ... 20
Figure 9 - Proposed pathway for oxidative metabolism of CBzM. (26) ... 21
Figure 10 - Structure of 1,3-diaryltriazenes. ... 23
Figure 11 - Synthesis of 1,3-diaryltriazenes and their N-acyl derivatives. ... 23
Figure 12 - Diminazene aceturate structure and physicochemical properties. (30) ... 24
Figure 13 - Example of alkyne analogue of DMZ. ... 26
Figure 14 - Sonogashira coupling. Adapted from (36). ... 26
Figure 15 - Structure of compound 11 and its rationale. ... 27
Figure 16 - 1,3-diaryltriazene with anti-mycobacterial activity. ... 28
Figure 17 - Triazenide salts 13a-13d. Adapted from (31). ... 29
Figure 18 - BaP metabolization catalyzed by CYP. ... 31
Figure 19 - Resveratrol. ... 31
Figure 20 - Compound 15. ... 32
Figure 21 - Synthesis of Compound 15. Adapted from (43). ... 32
Figure 22 - Examples of triazenoheterocycles. ... 34
Figure 23 – Compound 20. ... 35
Figure 24 - Degradation of SMA41 and BJ2000. Adapted from (60). ... 38
Figure 25 - Action of SMA41 and BJ2000. Adapted from (60). ... 39
Figure 26 - Hydrolysis and binary targeting of ZRBA1. (61) ... 40
Figure 27 - JDE52. (64) ... 41
Figure 28 - Chemical decomposition and targets of RB24. Adapted from (65). ... 42
Figure 29 - RB107. ... 42
Figure 30 - ZRS1. ... 43
Figure 31 - Synthesis of SMA41, BJ2000 and ZRBA1. Adapted from (74). ... 44
Figure 32 - Synthesis of RB24 and RB107. Adapted from (66). ... 44
Figure 33 - Synthesis of ZRS1. Adapted from (71). ... 45
Figure 34 - Imatinib and Dasatinib molecules. ... 46
Figure 35 – ZRCM5 and its hydrolysis. Adapted from (65). ... 46
Figure 36 - ZRF1. Adapted from (78). ... 47
Figure 37 - Synthesis of Compound 23. ... 47
Figure 38 - Synthesis of Compound 26. ... 48
Figure 39 - Synthesis of ZRMC5 and ZRF1. Adapted from (80). ... 48
Figure 40 - Compound 27 and its hydrolysis. Adapted from (65) ... 49
Figure 41 - Synthesis of Compound 27. Adapted from (82) ... 50
Figure 42 - Examples of first, second and third generation PARP inhibitors. ... 51
Figure 43 - EG22 and its hydrolysis. Adapted from (86) ... 52
Table Index:
Table 1 - Structures of some acyltriazenes. Adapted from. (14) ... 18
Table 2 – Advantages of DMZ bind tightly to G-quadruplexes DNA. ... 26
Table 3 - Extrahepatic organs where CYP1A1 and 1B1 are expressed. ... 30
1 Introduction
1.1 Triazenes in anticancer chemotherapy
Cancer is a multifaceted disease and represents one of the leading causes of mortality in developed countries exceeded only by heart diseases. The treatment of cancer, in particular metastases, is still one of the many challenges of medicine. (1)
Radiation therapy became a valuable tool after 1960 and, before that, just surgery was mainly used. The beginning of the modern era of chemotherapy is directly related with the discovery of nitrogen mustards as an effective treatment and with the necessity to eradicate metastatic cancer that surgery or radiation could not. Drugs, biological molecules and immune-mediated therapies have therefore become the focus for the current efforts to cure cancer and the target-therapy revolution has arrived. The new targets included growth factors, signalling molecules, cell-cycle proteins, modulators of apoptosis and molecules that promoted angiogenesis. (2)
Triazenes are a very useful and diverse class of compounds that have been studied, principally, for their anticancer potential in many tumors including brain, leukemia and melanoma. They are straight-chain molecules that contain three contiguous nitrogen atoms, in which N1 is double-bonded to N2, which is linked by a single bond to N3 (e.g. R1N=N–NR2R3 - Figure 1) and their mechanism of action is based on the generation of an alkyldiazonium species that damages DNA at the O6 and N7 positions of guanine. These group of compounds have an excellent pharmacokinetic, limited toxicity and also have similar chemical, physical, antitumor and mutagenic properties. (3–5)
Triazene are easily synthesized from available anilines or alkyl azides (Figure 2). In the aniline way, the synthesis is performed with nitrite ion under acidic conditions to form a diazonium salt, which reacts with a primary or secondary amine. To obtain triazenes
from alkyl azides, a reaction of an alkyl azide with the appropriate Grignard or alkyl lithium reagent must occur. (3)
Triazene-derived compounds of high clinical interest are alkylating agents dacarbazine (DTIC) and temozolomide (TMZ). (6)
1.2 Dacarbazine
DTIC, i.e. 5-(3,3-dimethyltriazeno)4-carboxamide, is an imidazole-carboxamide derivate that belongs to triazene class of 1-aryl-3,3-dialkyltriazenes and is structurally related to purines. This compound was synthesized for the first time in 1959 as a result of a rational attempt to develop a drug able to interfere with purine biosynthesis. Chemically, is composed of an imidazole ring fused with an amidic group and with a mono unsaturated chain of three nitrogen atoms ending with two methyl groups (Figure 3). (5,7,8) Dacarbazine has been in clinical use for the treatment of malignant melanoma, soft-tissue sarcoma, and Hodgkin’s disease since 1970s, when DTIC was approved in USA and in France. (3,5)
Although DTIC shows features similar to an intermediate metabolite of purine biosynthesis, the principal mechanism of action of the agent does not allow to classify this molecule as an antimetabolite. DTIC is a prodrug that requires metabolic activation,
Figure 3 - Structure of dacarbazine.
principally in the liver, by cytochrome P450 isoforms (CYP1A1, CYP1A2 and CYP2E1) to give a hydroxymethylated compound (HMTIC). Then, HMTIC, by the loss of formaldehyde, is converted into its monomethyl derivate (MTIC) which has a very short half-time and decomposes spontaneously into an inactive derivative, 5-aminoimidazole-4-carboxamide (AIC) and methyldiazonium cation, the alkylating specie. This alkylating agent is responsible for producing methyl adducts in DNA. In quantitative terms, the most frequent site of DNA alkylation is the N7 position of guanine. Nevertheless, the alkylation on the O6 position is the principal responsible for the cytotoxicity and the mutagenic effect of DTIC because it can generate an incorrect base pairing with thymine (Figure 5). (5,7–9)
DTIC is administered by the intravenous route and the most common side effects are nausea and vomiting, myelosuppression, cardiac and hepatic toxicity, immune depression and mucocutaneous toxicity. (5,10,11)
1.3 Temozolomide
TMZ, i.e. 8-carbamoyl-3-methylimidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one, is an alkylanting agent of imidazotetrazine class that is structurally and functionally related to DTIC. It was first synthesized in 1984 and, chemically, is composed by an imidazole ring with an amidic group bound to C1 (imidazole-carboxamide) condensed with a second tetrazinone ring system, that contains three adjacent nitrogen atoms (Figure 4). Temozolomide was approved by FDA and EMEA in 1999 and, because of its good central nervous system distribution, was first used to treat both primary brain tumors like glioblastoma multiforme and oligodendroglioma, and to radiosensitise these tumors. Latter, TMZ proved its efficacy in the treatment of melanoma. (5,12)
In contrast to Dacarbazine, TMZ spontaneously hydrolyses to MTIC in aqueous solution at physiological pH with a non-enzymatic conversion. Thereafter, MTIC rapidly degrades to AIC and methyldiazonium cation. This process of conversion is irreversible and pH-dependent (Figure 5). (5,7)
Due to the fact that TMZ does not require metabolic activation by the liver, it is also active in the case of liver function impairment and has wide potential clinical applications including its use for loco-regional therapy. In addition, the intact TMZ molecule, but not Dacarbazine or MTIC, crosses easily the blood brain barrier, because of its lipophilic character, and is then activated in the brain compartment. Moreover, differently from DTIC, the drug can be successfully administered by both parenteral and oral routes because it is optimally adsorbed by the intestinal tract (100% bioavailability). Temozolomide is also associated with a low incidence of severe adverse events and the most common side effect is myelosuppression. In contrast, nausea and vomiting are limited (10-15%), whereas the same side effects are remarkably severe and highly frequent in patients treated with DTIC. Today, because of all these characteristics, TMZ is almost replacing Dacarbazine in the clinic. (5,7,11,13)
1.4 Goals of monography
The focus of this monography is the fundamental physicochemical properties of some triazenes, namely, prodrugs of aliphatic triazenes, 1,3-diaryltriazenes, aryl morpholino triazenes, triazenes heterocycles and combi-triazenes with emphasis on the mechanisms of their antitumor action and how can they be developed as effective antitumor agents. In general, the synthesis of these compounds is always explained as well as their advantages in anticancer therapy.
2 Materials and methods
This monography was carried out through a comprehensive analysis of articles and patents. The articles were searched in the following databases: b-on, ScienceDirect, PubMed and Web of Science. The search of patents was performed in the United States Patent and Trademark Office (USPTO).
The keywords used were “triazene”, “alkyltriazene”, “1,3-diaryltriazene”, “aryl morpholino triazene”, “triazenopyrazoles”, “triazenoindazoles”, “triazenopyrroles”, “triazenoindoles” and “combi triazenes”. All the references found during the conduction of this research were studied in detail and were considered valid the full-text articles which contain the keywords mentioned above in the title and/or abstract. The references of the selected articles were also investigated for additional information.
3 Prodrugs of aliphatic triazenes
Arylalkyltriazenes have been subject of study by chemists and biologists for many years due to the fact that many members of this class shown mutagenic, carcinogenic and antitumor properties. One of this triazenes, DTIC, as described above, has proven to be therapeutically valuable. (14,15)
Although the first preparation of alkyltriazenes was reported by Dimroth more than a century ago (16), this class of compounds remained essentially unstudied until the 80s. These compounds, di- and trialkyltriazenes, are also DNA-alkylating agents but, in contrast to the aryldialkyltriazenes, do not require metabolic activation due to their instability. These aliphatic triazenes are very sensitive to proteolytic decomposition and most of them are stable in aprotic media or as pure compounds but are rapidly hydrolyzed in water. (3,14–18)
Alkyltriazenes have been useful model compounds for the study of DNA alkylation in the absence of metabolic activation, however biological studies were complicated by the fact that they are extremely unstable in aqueous solutions. Therefore, in an effort to acquire greater chemical stability while retaining alkylating activity, the 1,3-dialkyl-3-acyltriazenes1 were developed. (14,19)
3.1 1,3-dialkyl-3-acyltriazenes
1,3-dialkyl-3-acyltriazenes are another class of triazenes which show potent antitumor activity and are more stable to proteolytic decomposition than the parent 1,3-dimethyltriazene. The general structure is represented in the Figure 6 and structures of some acyltriazenes are given in Table 1. (3,14,20)
1 The term 1,3-dialkyl-3-acyltriazenes refers to compounds with N3 directly attached to a carbonyl-containing a substituent and it is used for convenience to emphasize the chemical behavior resulting from the presence of the carbonyl moiety. It is noted that the substituents like carbethoxy or methylcarbamoyl are not acyl groups in the strictest usage of the term. (14)
Table 1 - Structures of some acyltriazenes. Adapted from. (14) Compound Abbreviation R R’ X 1a DMA CH3 CH3 CH3 1b DMC CH3 CH3 OCH2CH3 1c DMM CH3 CH3 NHCH3 1d CMC CH2CH2Cl CH3 OCH2CH3 1e CMM CH2CH2Cl CH3 NHCH3 1f CBzM CH2CH2Cl CH2C6H5 NHCH3 3.1.1 Synthesis
The simplest examples of acyltriazenes are those that, like DMA, DMC and DMM, contain a 1,3-dialkyl-3-acyl structure. In the synthesis of these compounds, except for DMM, the direct acylation of the triazene was very sluggish or would not proceed because of low nucleophilicity of dialkyltriazenes. Therefore, in the case of DMM the direct acylation is possible and 1,3-dimethyltriazene reacted smoothly and directly with methyl isocyanate in pentane. The compounds DMA and DMC, in turn, are obtained, respectively, from the reaction of an acid chloride or ethyl chloroformate with a disubstituted triazene anion. The disubstituted anions are obtained by treating an alkyl azide with the appropriate alkyl lithium or Grignard reagent followed by addition of potassium hydride. All of these reactions produce the compounds with good yield. (3,15,21)
Although the synthesis of simple 1,3-dialkyltriazenes and their acylated derivatives can be accomplished in a straightforward manner by the reaction of alkyl azides with alkyllithiums or Grignard agent, this route was not applicable to chloroethyl derivatives like CMC, CMM and CBzM. It is not possible to prepare organometallic reagents containing a heteroatom on the b-carbon because of immediate elimination and, similarly, attempts to add organometallic reagents to 2-haloethyl azides also resulted in elimination reactions. So, in order to prepare these chloroethyl derivatives a less direct
approach was adopted2 (Figure 7). The first compound, 2-azidoethanol 2, was prepared
by the reaction of 2-chloroethanol with sodium azide. Then, the reaction with tert-butyldimethylsilyl3 chloride (TBDMS-Cl) catalyzed by imidazole, results in the formation of the compound 3. The protected azide was then reacted with methyllithium in diethyl ether solution, using the reverse addition technique (methyllithium added to the azide) in order to minimize the possible reaction of the organometallic reagent with the protecting group. If the objective was to prepare the compounds 5a and 5b, the resulting product was converted, in diethyl ether to its anion by the reaction with slight excess of potassium hydride, catalyzed by 18-crown-6 ether and then, the resulted solution was treated with ethyl chloroformate to give compound 5a or with acetyl chloride to give compound 5b. In the case of compound 5c, the formation of the anion compound is not necessary. Finally, the deprotection of the siloxytriazenes was accomplished with tetra-n-butylammonium fluoride in tetrahydrofuran solution and then, the triazenes were converted smoothly to the chloroethyl derivates by reaction with carbon tetrachloride and triphenylphosphine. (22,23)
2 This method can be applied to all chloroethyl derivates including CBzM with the necessary modifications.
3 The tert-butyldimethylsilyl (TBDMS) group is used widely for the protection of hydroxyl group in synthetic organic chemistry due to their ease installation and removal without affecting other functional groups. (88)
3.1.2 Mechanism of action
Acyltriazenes also decompose chemically to yield alkyldiazonium ions, but with more complex mechanism. Generally, they exhibit acid-catalyzed decomposition at lower pH, uncatalyzed decomposition at neutral pH, and base-catalyzed decomposition at higher pH. (14,20) The mechanism of action of most of these compounds at acidic or neutral pH involves N2–N3 heterolysis (Major Path A), leading to production of an alkyldiazonium ion derived from the N1 alkyl group and an amide derived from the N3 portion of the molecule. The alkyldiazonium ion can either chloroethylate (Minor Path D) or hydroxyethylate DNA (Major Path C). At basic pH, or in the presence of a hydrolytic enzyme such as esterase, deacylation becomes the predominant pathway leading to the prodution of two tautomeric dialkyltriazenes (Minor Path B), which can then decompose to give either a chloroethylating or hydroxyethylating species, or a methylating agent. The different ratios of methylation to chloroethylation or hydroxyethylation depend on the triazene structure (Figure 8). (20,24,25)
The biological activity of acyltriazenes and especially of acyltriazenes bearing a carbethoxy or a carbamoyl substituent could not easily be explained by chemical decomposition alone due to their high degree of chemical stability under physiological conditions. Therefore, an important question concerning the ability of these compounds to alkylate DNA in vivo was raised. A partial answer to this question came from findings that in the presence of porcine liver esterase, the rate and extend of alkylation by CMC were increased markedly and thus, it was postulated that CMC and similar compounds would be potent alkylating agents in vivo, but via a prior enzymatic deacylation step. However, during the course of the investigations, CMM exhibited much less toxicity than its corresponding carbethoxytriazene analog, CMC. So, the great disparity in their toxicity led to the hypothesis that their behaviour in vivo must be strikingly different. Due to the fact that CMC and CMM differ only in the acyl substituent, studies were consequently initiated to determine the metabolic pathway of the (methylcarbamoyl)triazenes, focusing on CBzM as a model compound. (14,26,27) Results indicated that degradation of the (methylcarbamoyl)triazenes was oxidative rather than hydrolytic and was most likely catalysed by one or more isozymes of cytochrome P450. Further results proposed that the (methylcarbamoyl)triazenes, CBzM
Figure 9 - Proposed pathway for oxidative metabolism of CBzM. (26) 1f
and CMM, are subject to oxidative metabolism at a minimum of two sites: methylcarbamoyl and chloroethyl substituents as shown in Figure 9.
Hydroxylation of the methylcarbamoyl group may lead to demethylation, at least in the case of CBzM, but does not directly result in breakdown of the triazene or the generation of an obvious alkylating agent. In contrast, hydroxylation of the chloroethyl group results in destruction of the triazene moiety, with formation of chloroacetaldehyde, a substituted urea, and presumably molecular nitrogen.
Therefore, the last investigations indicated that the antitumor efficacy of the (methylcarbamoyl)triazenes may be attributable to a unique mechanism that does not involve hydrolytic deacylation or the formation of an alkyldiazonium ion. However, the biological activity of these compounds may be explained with the formation of chloroacetaldehyde, a known mutagenic DNA alkylating agent. (26)
4 1,3 – diaryltriazenes
Aryl triazenes are a class of compounds that have been studied over 130 years for their interesting structural, anticancer, and reactivity properties. 1,3-diaryltriazenes belongs to this class and chemically are constituted of two aromatic groups connected by a triazene bridge (Figure 10). (28–30)
4.1 Synthesis
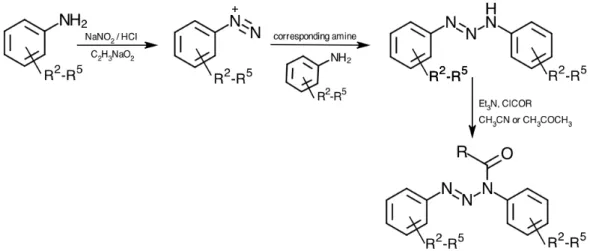
The general synthesis of 1,3-diaryltriazenes and N-acyl derivatives via diazonium intermediates is represented in Figure 114. The first step, is done by treating the appropriately substituted anilines with concentrated hydrochloric acid (HCl), sodium nitrite (NaNO2) and saturated sodium acetate (C2H3NaO2) at low temperature. The second step, consists in the addition of the corresponding amine, if the final compound is not symmetrical. The last and optional step relative to acylation of the selected 1,3-diaryltriazenes into N-acyl-1,3-1,3-diaryltriazenes, is achieved by using the appropriate acyl chloride (ClCOR) in acetonitrile (CH3CN) or acetone (CH3COCH3) solution in the presence of triethylamine (Et3N) as a base. (6,30–32)
4 R2-R5 represent the various substituents that can be added.
Figure 11 - Synthesis of 1,3-diaryltriazenes and their N-acyl derivatives. 8
4.2 Mechanism of action
4.2.1 Diminazene aceturate (Berenil) and derivatives
The well-known representative of 1,3-diaryltriazenes is diminazene aceturate (BerenilÒ or DMZ), the salt of 1,3-bis(4-amidinophenyl) triazene (Figure 12). This compound is an aromatic diamidine of synthetic origin that was developed more than six decades ago. BerenilÒ has been in the market since 1955 for the control of trypanosomiasis caused by several species of flagellated protozoa belonging to the genus Trypanosoma that are responsible for a large number of infections in animals. In addition to its trypanocidal activity, diminazene aceturate has also demonstrated applicability in the treatment of animals infected with protozoa of the genus Babesia. (6,30,33)
The capacity of diminazene aceturate to bind to DNA has been recognized very early. (34) Because of its chemical structure that contains two identical cationic groups (dicationic diamidine), this compound presents a great affinity for the sequences of adenine-thymine base pairs. Therefore, the binding to DNA occurs via complexation
Figure 12 - Diminazene aceturate structure and physicochemical properties. (30) 9
into the minor groove of AT-rich5 domains of DNA double helices. DMZ can also bind
to RNA and to DNA duplexes, exhibiting characteristic properties of both intercalation as well as minor groove binding. (6,30) Nevertheless, recently it was revealed that diminazene actually binds to G-quadruplexes 1000 times better than DNA duplexes, with dissociation constants approaching 1 nM. (35,36)
G-quadruplexes are secondary structures in both DNA and RNA that are emerging as important regulatory elements that control diverse processes in the cell, ranging from telomere maintenance, gene expression, translation, alternative splicing, RNA metabolism and protein sequestration.
There are ~3 760 000 guanine-rich regions in the human genome, which have the potential to form G-quadruplexes including those at the telomere end and promoter regions. In animal chromosomes, the telomerase enzyme (which is up-regulated in certain cancers) is responsible for maintaining the telomere length thereby rendering cancer cells immortal. The telomere is G-rich and has been shown to be capable of forming G-quadruplexes. In addition to telomeres, G-quadruplexes are present in the promoter regions of a number of oncogenic genes such as myc, BCL-2, KRAS and c-kit. Small molecules that stabilize the G-quadruplex structure have been shown to inhibit the extension of the DNA substrate by telomerase and transcription of cancer-related genes and hence these molecules have the potential to be used as anticancer agents.
Nevertheless, DMZ does not have strong anticancer activity because the binding of this compound to the minor groove of AT-rich DNA with a micromolar dissociation constant would limit the targeting of G-quadruplexes. In fact, in the complex cellular environment the concentration of duplex DNA is several orders of magnitude greater than G-quadruplexes. Despite this, the advantages of DMZ listed in the Table 2, shows that DMZ scaffold is a good starting point to develop potent G-quadruplexes ligands. So, recently, it was discovered that alkyne analogues of DMZ that also bind to G-quadruplexes have good anticancer properties against ovarian, prostate and triple negative breast cancers. (35,36)
5 AT-rich are regions with high content of adenine and thymine residues. This repeated sequences are commonly present in the sites for DNA replication initiation in bacterial, archaeal, and eukaryotic replicons. (89)
Table 2 – Advantages of DMZ bind tightly to G-quadruplexes DNA.
1. Connections with other biomolecules are not probable because DMZ
does not readily form p-aggregates.
2. The amidine groups on DMZ improve aqueous solubility and it is an
important factor for drugs.
3. DMZ has a simple structure and could be readily diversified and
synthesized on a large scale.
4. The amidine group, which is protonated at physiological pH, also would
facilitate drug permeation across lipid membranes.
These compounds, monoamidine analogues that bear alkyne moieties are, unlike of DMZ, selective G-quadruplex binders with good anticancer properties. This occurs due to the fact that since DMZ requires both amidine groups to bind to duplex DNA, removing one amidine group will reduce duplex DNA binding affinity. One example of these compounds is shown in the Figure 13.
The synthesis of this compounds is shown in Figure 11. The corresponding amines, in the case of the example of the figure above, alkyne-substituted aromatic amines, are synthetized via Sonogashira coupling (Figure 14). (36)
Figure 13 - Example of alkyne analogue of DMZ.
Figure 14 - Sonogashira coupling. Adapted from (36). 10
4.2.2 4-nitro-substituted 1,3-diaryltriazenes
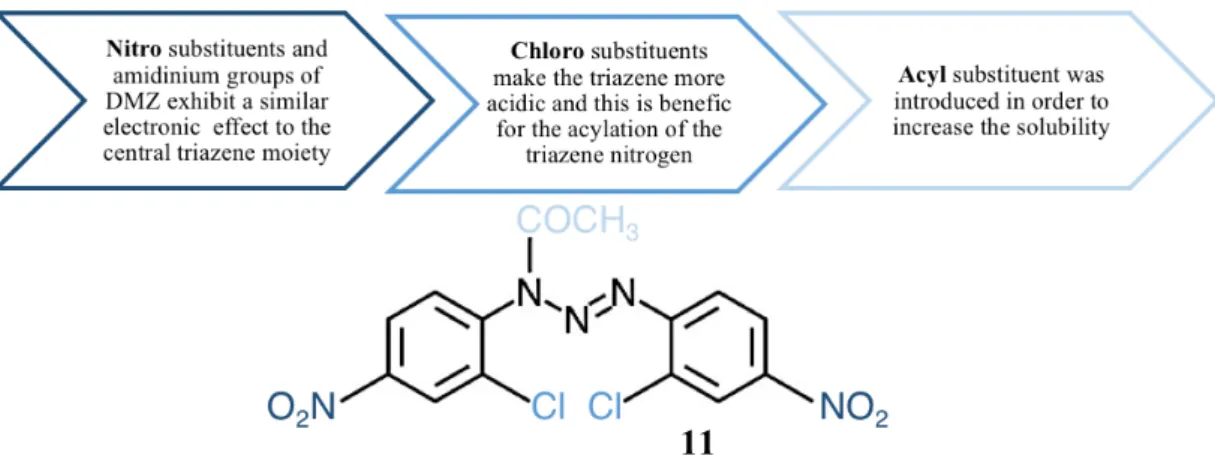
As mentioned before, diminazene aceturate has very low activity against tumor cells (IC50>100 µM). Therefore, in order to improve the antitumor efficacy of diarytltriazenes, it was demonstrated that inactive 1,3-diaryltriazenes can be modified to highly cytotoxic compounds by the introduction of two nitro groups at the para position of the benzene rings, and two additional electron-withdrawing groups (bromo, chloro, trifluoromethyl, or fluoro substituent) at their ortho positions. These compounds, 1,3-bis(4-nitrophenyl)triazenes, show cytotoxicity at very low concentrations (IC50 0.22 to 12.8 µM). (6,37,38) he compound 11, 3-acetyl-1,3-bis(2-chloro-4-nitrophenyl)-1-triazene, shows highly cytotoxic activity against different tumor cell lines, including cisplatin-resistant laryngeal carcinoma cells. Notably, its antiproliferative action is significantly higher against tumor cells than against normal cells. The rationale behind the structure of this compound and the structure itself are represented in the Figure 15.
The mechanism of action of this compound is not related with the binding to DNA, unlike the alkyltriazenes (dacarbazine and temozolomide) or the diaryltiazene derivative, Berenil. Instead, compound 11 induces the formation of reactive oxygen species (ROS) and the endoplasmic reticulum (ER) stress response, resulting in apoptosis. Moreover, recently, the cytotoxicity was also associated to the induction of the SAPK/JNK signalling pathway. (6,38)
11
4.3 Other therapeutic interests
In addition to the already mentioned antitumor and antitrypanosomal activities of these compounds other therapeutic interest have arisen, such as anti-mycobacterial (39) and antibacterial (31) activities.
4.3.1 Anti-mycobacterial activity
Identification and development of novel compounds to target Mycobacterium tuberculosis, the etiological agent of tuberculosis (TB), is still of utmost importance mainly due to the rapid generation and spread of resistances. For this reason, several substituted 1,3-diaryltriazenes were evaluated for its potential as anti-tubercular agent. The compound 12 represented in the Figure 16 was selected with the best biological properties (IC50 = 3.26 µM). This compound showed the ability to inhibit the growth of a multi-drug resistant M. tuberculosis as well as the intracellular replication of M. tuberculosis.
Despite these good results, it is unlikely that these compounds could find their application as first line drugs. This occurs because the TB chemotherapy requires intensive treatment for several months and the anti-tubercular potency of these compounds was accompanied with an acute cytotoxicity, combined with the DNA binding properties of the anti-trypanosomal drugs. However, with the increase in incidence of multi-, extensively- and total-drug resistant TB, when other treatment options are exhausted, this compound class might prove of use. (39)
4.3.2 Antibacterial activity against MRSA
Antibiotic resistance has become one of the most serious health care problems in the world and there is an urgent need to develop new effective antibacterial agents that circumvent the emergence of resistance.
The antimicrobial properties of 1,3-diaryltriazenes depended on the type of substituent group attached to the two constituent benzene rings. Specifically, a trifluoromethyl
Figure 16 - 1,3-diaryltriazene with anti-mycobacterial activity. 12
group at ortho position, and a nitro substituent at para position relative to the triazene was found to be critical to its activity. Thus, the triazenide salts represented in the Figure 17 showed high activity against one of the most clinically important bacterial species, MRSA.
These triazenide salts were prepared from the corresponding diaryltriazene. The reaction of the diaryltriazene with methyl (HCºCCO2CH3) or ethyl propiolate (HCºCCO2C2H5) in the presence of trimethylamine (Et3N) yielded compounds 13a and
13b, whereas trituration with trimethylamine (Et3N) and potassium hydroxide (KOH) gave triazenide salts 13c and 13d respectively.
The molecular basis of the triazene activity against S. aureus remained unclear and appears to have more than one mechanism of action. Although, it seems that occurs a modification of phospholipid metabolism and consequently the characteristics of the staphylococcal cell membrane.
In addition, the selected compounds were found to be very effective against other gram-positive bacteria, such as S. pneumoniae, B. subtilis, Vancomycin resistant E. faecalis, and M. smegmatis. (31)
5 Aryl morpholino triazenes
Cytochromes P450 (CYP) constitute a large family of hemoproteins involved in the detoxication of foreign compounds and the biosynthesis of endogenous compounds, including steroid hormones, bile acids, and cholesterol. More than 21,000 CYPs have been identified to date, and 18 families, including over 50 enzymes are found in humans.
P450 families 1, 2, 3, and 4 contribute most extensively to the transformation of xenobiotics to more polar metabolites that can be better excreted. The CYP1 subfamily contains three members: CYP1A1, CYP1A2 and CYP1B1. Human CYP1B1 share 41 and 40% amino acid sequence homology with human CYP1A1 and CYP1A2, respectively, while the latter two are 72% identical. CYP1A2 is expressed mainly in liver, whereas CYP1A1 and 1B1 are expressed in many extrahepatic organs (Table 3). (40,41)
Table 3 - Extrahepatic organs where CYP1A1 and 1B1 are expressed.
CYP1A1 Pancreas, thymus, uterus and small intestine
CYP1B1 Breast, prostate and uterus (manly)
Kidney, intestine, eye and brain (low levels)
Within CYP1 subfamily, CYP1A1 and CYP1B1 have been widely studied because they are involved in the conversion of a large number of polycyclic aromatic hydrocarbons (PAHs) into carcinogens. In fact, PAHs, found in tobacco smoke, automobile exhaust, and charbroiled meat, can be oxidized by CYP1A1 and CYP1B1 into carcinogenic epoxydiols through three enzyme-mediated reactions:
1. Oxidation of a double bond catalyzed by P450 enzymes to unstable arene oxides;
2. Hydrolysis of the arene oxides by microsomal epoxide hydrolase (EH) to dihydrodiols;
3. CYP-catalyzed oxidation at double bond adjacent to the diol function to generate a high reactive vicinal diol-epoxide.
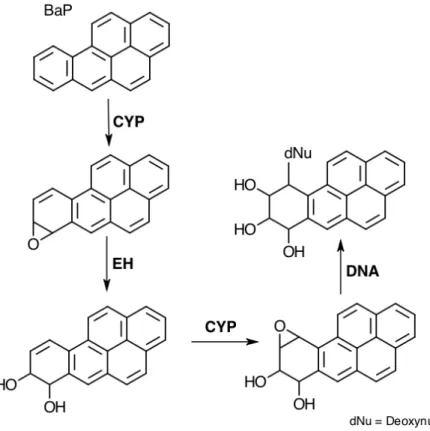
The formed epoxydiols are good electrophiles that react with the primary amino groups in adenosine and guanosine to form 2’-deoxyguanosine and 2’-deoxyadenosine in DNA. These DNA adducts cause misreplication, which lead to mutations in DNA that can cause cancer. Benzo[a]pyrene (BaP) is an example of PAH and its metabolization is given in the Figure 18. (42–44)
Because CYP1A1 and CYP1B1 oxidation of PAHs has been linked to tumor formation, various compounds have been studied for their ability to inhibit these enzymes such as stilbenes, naphthoquinone and anthraquinone derivatives, flavonoids, coumarin derivatives, alkaloids, and other compounds. Many of these inhibitors share several structural motifs: they are planar and contain one or more hydrophobic aromatic rings. One example is resveratrol (Figure 19), a stilbene molecule, that is planar with two aromatic rings and exhibit anti-cancer activity. (41,43)
Figure 18 - BaP metabolization catalyzed by CYP.
Figure 19 - Resveratrol. 14
Aryl morpholino triazenes have similar structural features and in addition contain a triazene unit that could form p-p interactions with the enzyme active site amino acids. Lee et al prepared several aryl morpholino triazenes and the compound 15, 4-[(E)-2-(3,4,5-trimethoxyphenyl)diazenyl]-morpholine (Figure 20), was the only compound that inhibited both CYP1A1 and CYP1B1 as well as or more so than resveratrol. Its IC50 was 10 µM with CYP1A1 and 18 µM with CYP1B1 compared to resveratrol’s IC50 values which were determined to be 34 µM with CYP1A1 and 55 µM with CYP1B1. These values indicate that the trimethoxy groups, which are electron donating and highly hydrophobic, provide a favorable interaction with the hydrophobic enzyme active site.
The compound 15 was synthesized at 0ºC by combining a mixture of 3,4,5-trimethoxyaniline and 6 M hydrochloric acid (HCl) with an aqueous solution of sodium nitrite (NaNO2) to produce a diazonium salt that underwent coupling with morpholine (Figure 21). (43)
In conclusion, aryl morpholino triazenes are a new class of compounds that inhibit CYP1A1 and CYP1B1 at the micromolar level. (43)
Figure 20 - Compound 15. 15
6 Triazenoheterocycles
As mentioned above, a triazene derivative currently used in anticancer therapy is dacarbazide. The potent antitumor activity shown by this compound led to the development not only of aryl but also of heteroaryl derivatives, such as triazenopyrazoles, -indazoles, -pyrroles and -indoles. (45,46)
6.1 Synthesis
The general synthesis of these compounds, like for other triazenes, was performed in two steps. The first step consists in the formation of the diazonium salt from the corresponding amine using an acid medium and aqueous sodium nitrite at low temperature. The second and last step, involves the addition of the appropriate amine in order to obtain the triazene compound. (46,47)
6.2 Properties
Triazene activity can be modulated by carrier structure, which influences pharmacokinetics and/or compound stability. So, the nature of the heterocyclic portion plays an important role and the activity increases as the rings become more electron-rich, although at the expense of stability. (47–49) Consequently, triazenoindoles are quite more active than triazenopyrroles and the same occur relatively to triazenopyrroles and triazenoindazoles. All of these compounds showed cytotoxic activity but, differently by Dacarbazine, did not follow a mechanism of action based on the microsomal activation. (45,50)
Several examples of these compounds are shown in the Figure 22. The triazenoindazoles (compound 17) showed more cytotoxic activity than the corresponding pyrazole derivatives (compound 16). The compound of the type of compound 17 with more activity was the diethyltriazenoindazole with chloro in the position 5 (IC50 = 11.7 µM against human leukemia cell lines). (50) In accordance with these results, 3-triazenopyrrole (compound 18), also showed cytotoxic activity against erythroleukemia cells in the range of 1.1-3.1 µM. However, the benzocondensation on this series that led to 3-triazenoindoles (compound 19) shown 20-40-fold more activivity than the pyrrole derivatives against erythroleukemia and multidrug-resistant cells with IC50 values of 0.053-0.080 µM and 0.10-0.14 µM, respectively. (46,49,51)
6.3 2-Triazenoazaindoles and pancreatic cancer
The majority of malignant tumors affecting the exocrine pancreas are histologically defined as pancreatic ductal adenocarcinomas. This type of tumor rapidly grow and metastasize representing one of the leading causes of cancer-related death in developed countries.
Current therapeutic treatments for patients with advanced disease show only modest effectiveness and are associated with considerable toxicity. Moreover, pancreatic cancer shows a considerable drug resistance. Overexpression or aberrant activation of members of the ErbB family of transmembrane tyrosine kinase growth factor receptors, which includes EGFR (ErbB1) and HER-2 (ErbB2), occurs frequently and is associated with multiple drug resistance and decreased patient survival.
In order to targeting these receptors, a novel low-molecular weight agent was developed, i.e. ethyl 2-(3,3-dibenzyl 1-triazenyl)-1H-pyrido(2,3-c)pyrrolo-3-carboxylate (Figure 23). Compound 20 induces decreased EGFR and HER-2 expression by blocking transcriptional genes and causes significant reduction of cell growth and metabolic activity in four human pancreatic cancer cell lines.
The observed biochemical selectivity of the compound 20 for both receptors together with its potent anti-proliferative effects makes the class of 2-triazenoazaindoles
Figure 22 - Examples of triazenoheterocycles.
16 17
attractive in order to produce novel chemotherapeutic agents for the treatment of patients with pancreatic cancer. (52)
Figure 23 – Compound 20. 20
7 “Combi-triazenes”
7.1 Development of resistance and concept of hybrid drugs
One of the reasons for failure of chemotherapy is development of resistance. Failure of a patient’s cancer to respond to a specific therapy can result from host factors listed in the Table 4 and specific genetic or epigenetic alterations in the cancer cells. This genetic and epigenetic heterogeneity in the face of the powerful selection imposed by potent anticancer drugs result in an overgrowth of drug-resistant variants and the rapid acquisition of resistance by many cancers.
Table 4 - Host factors.
1. Poor absorption or rapid metabolism or excretion of a drug, resulting in
low serum levels.
2. Poor tolerance to effects of a drug, especially elderly patients, resulting
in a need to reduce doses below optimal levels.
3.
Inability to deliver a drug to the site of a tumor, as could occur with bulky tumors or with biological agents of high molecular mass and low
tissue penetration such as monoclonal antibodies and immunotoxins.
4. Various alterations in the host-tumor environment that affect response of
the tumor including local metabolism of a drug by non-tumor cells.
5.
Unusual features of the tumor blood supply that may affect transit time of drugs within tumors and the way in which cells in a cancer interact
with each other and with interstitial cells.
Combinations of drugs that impact multiple targets simultaneously are the standard of care in cancer treatment as they are better at controlling complex disease systems and, more important, are less prone to drug resistance. In order to improve the efficiency of using a two-drugs cocktail, one approach involves the use of so-called hybrid drugs, which comprises the incorporation of two drugs in a single molecule with the intention of exerting dual drug action. (53)
The first approach that has been used to design new anticancer hybrids is based on the ability of a combination of haptophoric moieties on a new molecular structure to retain their affinity and activity for the biological targets. This concept is achieved using two strategies:
1. Merging of two haptophoric groups selected from two drugs exhibiting the same cytocidal mechanism of action with the aim of improving activity, selectivity and biopharmaceutical properties.
2. Merging of haptophoric groups from two drugs acting through different mechanisms of action to improving the pharmacokinetic and pharmacodynamics properties of the parent components as well as to synergize their mechanisms of action in a single molecular entity structure.
The second approach combines two or several entire drugs in the same molecular structure (combi-molecules) with similar or different mechanism of action. The connection of the two molecular entities can be achieved using cleavable or non-cleavable linkages. While the connection through non-non-cleavable linking arms is based on the ability of the different molecules to retain their biological activity and their specific and respective affinity for their biological targets, the approach using cleavable bond is based on the release of two parental molecular structures under physiological or the enzymatic conditions to improve pharmacokinetic and selectivity. (54)
7.2 EGFR TK inhibitor-linked DNA damaging agents
7.2.1 First generation of combi-molecules
Initially the feasibility of the combi-molecule strategy was proven by the first prototype of combi-molecules SMA41 and BJ2000.
Solid tumors are often characterized by the expression of DNA repair enzymes, that confer resistance to chemotherapy, and by the overexpression or dysfunction of proteins directly implicated in mitogenic signaling. The expression of DNA repair enzymes, such O6-alkylguanine-DNA alkyltransferase (AGT), significantly decreases the chemosensitivity of these tumors to alkylating agents like triazene class. On the other hand, the overexpression and dysregulation of tyrosine kinase (TK) is commonly observed in a large number of cancers and is associated with aggressive tumor progression, poor patient survival and more important reduced chemosensitivity. For
example, the epidermal growth factor receptor (EGFR) is often overexpressed in breast, ovarian, and prostate tumors. (55–58)
The inhibitors of EGFR tyrosine kinase that belong to the quinazoline class act by competitively binding to its ATP binding site, thereby blocking subsequent activation of downstream signalling cascades, including mitogen-activated protein (MAP) kinase and the transcription of genes associated with cell proliferation. Due to the fact that this type of drugs required prolonged and repeated doses for the induction of sustained antitumor activity, the combination of this drugs with classical cytotoxic agents has become a common approach to increase the potency of EGFR-directed therapy. (58,59) The “combi-triazenes” SMA41 and BJ2000 are both a chimeric and unimolecular combination of two molecules associated with two major mechanisms of action: 4-anilinoquinazoline that defines its ability to inhibit EGFR-mediated cell signalling and 3-methyl-1,2,3-triazene that masks its alkylating metabolite. These chimeric molecules are able to target EGFR on its own and to degrade under physiological conditions to give SMA52/FD105 (inhibitors of EGFR – I) and methyldiazonium (an alkylating agent – TZ) as shown in the Figure 24. The cytotoxic contribution of the methyldiazonium to DNA damage depend not only on the O6-alkylguanine adducts, but also on the N7-methylguanine adducts what are important to minimize problems associated with the presence of AGT. (57–60)
As depicted in the Figure 25, TZ-I can penetrate in EGFR-overexpressing cells by passive diffusion and once inside the cells either directly bind to the EGFR ATP-binding site to provide the TZ-I-EGFR complex (path 3) or degrade into a cytotoxic TZ molecule plus an EGFR TK inhibitor I (path 2). More importantly, the TZ-I may directly alkylate the EGFR, as outlined in path 4, wherein an inactivated (covalently
modified) receptor (TZ-EGFR) may be formed, leaving an irreversibly inhibited receptor. (59,60)
The only difference between SMA41 and BJ2000 is that the latter, with a less bulky chloro substituent, has 2-fold stronger affinity than SMA41 and is capable of generating an EGFR TK inhibitor with 5-fold stronger affinity than SMA52. (60)
7.2.2 Combi-molecules with improved properties
SMA41, the first prototype combi-molecule studied in vivo, showed a rather moderate antitumor activity and this was imputed principally to:
1. Poor water solubility;
2. Decreased potency observed in AGT-expressing cells; 3. Very short half-life.
In an attempt to develop a better approach, a new prototype combi-triazene, ZRBA1, was synthesized. This new molecule contain a 3’-chloro group in the quinazoline ring because, as mentioned above, BJ2000, with chloro group showed superior potency, and a N,N-dimethylaminoethyl group attached to the N3 of 1,2,3-triazene. The last group improved hydrosolubility because its polar properties promotes aqueous solvatation and, more importantly, induce N,N-dimethylaminoethylguanine adducts that are perhaps less susceptible to AGT repair than those produced by SMA41. Moreover, ZRBA1 demonstrated improved stability than SMA41 with longer half-life (108
minutes compared with 30 minutes) maybe due to its ability to form intramolecular hydrogen bonding that stabilizes the conjugated tautomer (Figure 26). (61)
7.2.2.1 ZRBA1 as a radiosensitizer
In locally advanced solid tumors, the combination of cytotoxic treatments such as chemotherapy and radiation has been shown to improve local control, organ preservation, and long-term survival.
ZRBA1 has radiosensitizer potential that may be secondary to its ability to arrest the cells in G2/M, a cell cycle phase in which tumor cells are sensitive to radiation. The combination of this molecule with radiation increased levels of DNA damage that associated with the concomitant downregulation of EGFR-mediated signaling by ZRBA1 contribute for significant levels of cell killing and can enhance tumor response. (62,63)
7.2.2.2 JDE52: a bistriazene combi-molecule
JDE52, a bistriazene combi-molecule, was designed with the aim of improve the potency of ZRBA1. Like is shown in the Figure 27, this bistriazene was programmed
to release two equivalents of EGFR TK inhibitor FD105 and a more cytotoxic bifunctional DNA-damaging species. In fact, apoptosis was triggered by JDE52 at a faster rate than ZRBA1 and led to higher levels of cell killing. This superior potency of JDE52, when compared with ZRBA1, may be imputed to mechanisms associated with the generation of higher intracellular concentrations of FD105 and to the induction of DNA cross-links. (64,65)
7.2.3 Cascade-release targeting combi-molecules
In order to enhance the potency and stability of the combi-targeting molecule a novel strategy termed “cascade release” (CR) was developed. This strategy seeks to mask the combi-molecule into a prodrug planned to release the antitumor species by hydrolytic activation.
RB24, a masked methyltriazene, was designed according with the premise that acetoxymethyltriazenes are known to be hydrolyzed to the hydroxymethyltriazene intermediate that rapidly degrades into the corresponding monoalkyltriazene and this further heterolyses latter to an aromatic amine plus a DNA-damaging species. Thus, RB24 degrade to RB14, ZR08, RB10 and a DNA alkylating methyldiazonium ion in a
progressive way, as shown in the Figure 28. In this cascade, in addition to methyldiazonium ion, another reactive electrophile is formed: an iminium ion. This ion alkylates the active site of EGFR, thereby irreversibly blocking its action.
RB24 induced significantly higher levels of DNA damage in EGFR transfected cells than in wild type cells what indicate that RB24 could selectively target the cancer cells overexpressing EGFR. Moreover, RB24 showed great antiproliferative activity that could be imputed to the additional growth factor receptor TK irreversible inhibitory property imprinted in the molecule that induced a depletion of DNA repair protein and inactivation of anti-apoptotic signaling while high level of cytotoxic DNA lesions were being inflicted. (65–69)
RB107 (Figure 29) is also a cascade release molecule with an acetoxymethyl function that is hydrolyzed rapidly to generate BJ2000, a monoalkyltriazene that further degrades to FD105 and DNA alkylating methylating species. This molecule has similar properties to RB24. (70)
Figure 28 - Chemical decomposition and targets of RB24. Adapted from (65).
7.2.4 Combi-molecule containing N-acetoxymethyl carbamate
Previous strategies for stabilizing combi-triazenes were based on masking 1,2,3-triazene chain with 3-acetoxymethylene group as in RB24 and RB107. While the latter type of molecules were more stable than their parent triazenes, their half-lives were only 5-10 minutes longer than the latter in cell culture medium. In order to further increase the bioavailability of combi-molecules, a novel approach that seeks to mask the monoalkyltriazene with carbamates was designed and the half-lives were prolonged 20-55 minutes. (70–72)
ZRS1 (Figure 30), that contains a more stable acetoxymethyl carbamate function, is a second-generation derivative of RB107. This molecule, in vitro was extremely stable, but it is rapidly metabolized in vivo perhaps due to intracellular or plasma esterases that cleave the acetoxymethyl function. ZRS1 reached the plasma where rapidly releases BJ2000, which in turn decomposed to FD105 and the methyldiazonium species. A fraction of ZRS1 and BJ2000 may reach the tumors and decompose in situ. Plasma-released FD105 was further metabolized in the liver to give its acetylated metabolite FD105Ac that was further delivered to tumors. This FD105Ac metabolite was shown to be more potent EGFR inhibitor than FD105 and it is significant over previous methylating combi-molecules. (70)
7.2.5 Synthesis of EGFR TK inhibitor-linked DNA damaging agents
The synthesis of the EGRF TK-inhibitor-linked DNA damaging agents, including the last mentioned RB24, RB107 and ZRS1, is done sequentially.
Firstly, the compounds SMA41, BJ2000 and ZRBA1 was synthesized as described in the Figure 31. The first compound, 5-nitroanthranilonitrile was treated with a sulfuric acid (H2SO4)/formic acid (CH2O2) mixture. The resulting quinazoline was heated with phosphorus pentachloride (PCl5) to provide the chloro compound, which was treated
with substituted anilines (ArNH2). The nitro compounds, in the case of X=Br (RB10), was reduced with Fe in ethanol, however in the other cases, was reduced by catalytic hydrogenation. Lastly, the triazene moiety was formed using NOBF4 in acetonitrile (CH3CN) followed by addition of the corresponding amine and neutralization with triethylamine (Et3N). (73,74)
In order to synthesize compounds RB24 and RB107, the compounds RB10 and FD105 were also treated in acetonitrile (CH3CN) with NOBF4, but with the addition of a 10:1 mixture of formaldehyde (H2CO) / methylamine (MeNH2) and an alkalinization with K2CO3. The resulting compound was treated with acetic anhydride (CH3CO)2O in pyridine to obtain RB24 or RB107 (Figure 32). (66)
Figure 31 - Synthesis of SMA41, BJ2000 and ZRBA1. Adapted from (74).
Finally, ZRS1 was obtained by treatment of BJ2000 with chloromethyl chloroformate and pyridine in acetonitrile (CH3CN) at cold temperature to give ZRL4. Exchange of the Cl atom using potassium iodide (KI) in dry acetone, gave the corresponding iodomethyloxycarbonyl-methyltriazene, which was treated with silver acetate to obtain ZRS1 (Figure 33). (71)
7.3 Bcr-Abl TK inhibitor-linked DNA-damaging agents
A reciprocal translocation between chromosomes 9 and 22 produces a Philadelphia (Ph) chromosome which leads to the formation of the novel Bcr-Abl fusion gene. This fusion gene leads to the production of an abnormal tyrosine kinase protein that activates multiple downstream signalling pathways resulting in survival and proliferation of chronic myeloid leukemia (CML) cells. In fact, Ph chromosome is present in the leukemia cells of more than 95% patients with CML.
The blockade of the Bcr-Abl TK activity with Imatibin and Dasatinib (Figure 34), two potent inhibitors approved by FDA for the treatment of CML, has been proven to exhibit significant antitumor activity. However, the development of resistance and relapses were observed and novel therapy models have been designed like combi-molecules. TK Inhibitors-resistance mechanisms can include, for example, mutations and amplification of Bcr-Abl genes or drug efflux mechanisms. (65,75–77)
ZRCM5, the first prototype of combi-molecule designed to release an Imatinib analog, contain a 2-phenylaminopyrimidopyridine moiety targeted to Bcr-Abl kinase and a triazene tail capable of generating a methyldiazonium species. ZRCM5 is a hydroxymethyltriazene that does not require metabolic oxidation to generate the cytotoxic species, since following loss of formaldehyde leads to the formation of monoalkyltriazene, a hydrolabile species that generates the methyldiazonium cation upon hydrolysis (Figure 35).
As expected, ZRCM5 was found to be approximately 74-fold more potent than temozolomide, which might be due to its ability to simultaneously block Bcr-Abl and related DNA repair activity, while inducing significant DNA lesions in Bcr-Abl expressing leukemia cells. (4,65) However, although its capacity of inducing high levels of DNA damage, exhibited only weak Bcr-Abl inhibitory activity what lead to the
Figure 34 - Imatinib and Dasatinib molecules.
design of a novel compound, ZRF1, that contain a trifluoromethyl benzamide moiety for enhancing its Bcr-Abl potency. (78)
ZRF1 (Figure 36) was “programmed” to degrade into another inhibitor ZRF0 plus a methyldiazonium species and demonstrated a 1.6-fold greater Bcr-Abl TK inhibitory potency than Imatinib and 37-fold greater potency than ZRCM5. Molecular modelling studies confirmed the importance of the hydrophobic interaction mediated by the CF3 group with the ATP-binding site of TK. (78)
More importantly, the superior potency of ZRF1 over Imatinib was more pronounced in Bcr-Abl-positive cells coexpressing wild-type p53, which is available for transactivating apoptosis protein p21 and Bax. Thus, ZRF1 is the first ever multitargeted combi-molecule exerting a tandem targeting of Bcr-Abl mediated antiapoptotic signaling and activation of the DNA damage response pathway. (65,78)
The synthesis of compounds ZRCM5 and ZRF1 is also done sequentially. Firstly, to obtain the compound 23, the compound 21 was treated with N,N-dimethyl-formamide dimethylacetal to form compound 22, which, in turn, was treated with substituted phenylguanidine. The nitro group was reduced by catalytic hydrogenation (Figure 37). (79)
Figure 36 - ZRF1. Adapted from (78).
21 22 23
Then, the commercially available compound 24 was hydrolyzed under acidic conditions to give compound 25, which was chlorinated with SOCl2 to give compound 26 (Figure 38).
Finally, compound 23 was added to compound 26 in methylene chloride (CH2Cl2)/pyridine at 0ºC to give the nitro compound, which was reduced with Fe in ethanol to provide the amine compound. The triazene moiety was formed with nitrosonium tetrafluoroborate (NOBF4) in acetonitrile (CH3CN) followed by the addition of methylamine (MeNH2) or 1:30 mixture of methylamine (MeNH2) and formaldehyde (H2CO) to obtain ZRF1 or ZRMC5, respectively. Then the solution was neutralized with triethylamine (Et3N) (Figure 39). (80,81)
25
24 26
Figure 38 - Synthesis of Compound 26.
23 26
7.4 AGT inhibitor-linked DNA-damaging agents
The alkylation of O6-position of guanine in DNA is the major source of antitumor activity of DNA alkylating agents, such as dacarbazine and temozolomide. However, a DNA repair enzyme previously mentioned, O6-alkylguanine-DNA alkyltransferase (AGT), also called O6-methylguanine-DNA methyltransferase (MGMT), can repair the O6-lesion of guanine by transferring the O6-alkyl group to the active center at the Cys145 residue and restore normal DNA. AGT is a suicide enzyme that is degraded after accepting the lesion groups and its activity can only be recovered by its resynthesis. (65,82–84)
Due to the fact that the increasing of AGT level correlates well with the enhancement of tumor resistance to guanine O6-alkylating agents, a series of AGT inhibitors were synthesized as adjuvants to improve the chemotherapeutic effects. O6-benzylguanine (O6-BG) was the first potent AGT inhibitor that had entered clinical. O6-BG acts by transferring its benzyl groups to active site of AGT inactivating the enzyme. Unfortunately, the combination of O6-BG with guanine O6-alkylating agents only exhibited limited response in clinical trials. (65,82,83)
To overcome this weakness and based on the concept of combi-molecule, a novel approach containing the DNA methylating triazenes and the antiresistance agent O6 -BG in one molecule was designed. An important advantage of this strategy is that AGT-depletion induced by O6-BG release and formation of the methylating species occur simultaneously and in the same environment, ensuring optimal effect from DNA methylation. The selected combi-molecule was compound 27 (Figure 40) because it has an optimal half-life of approximately 23 min and was the most active (IC50 » 10 µM). This superior activity is due to the AGT depletion by O6-BG released from the parent molecule and its favorable penetration property. (65,82)
Figure 40 - Compound 27 and its hydrolysis. Adapted from (65) 27