Depart ment s of Neurology1, Pat hology2and Pediat rics3, Facult y of M edical Sciences, (FCM ) St at e Universit y of Campinas (UNICAM P), Campinas SP, Brazil: aM D, PhD and Assist ant Prof essor; bM D, M ast er in M edical Sciences; cFull Prof essor. Present ed in part at t he 4t h Congress of t he European Paediat ric Neurology Societ y, Baden-Baden, Germany. Eur J Paed Neurol 2001;5:A55-56. Support ed in part by FAEP - UNICAM P 973/01.
Received 25 M arch 2004, received in f inal f orm 24 June 2004. Accept ed 4 August 2004.
Dra. Anamarli Nucci - Depart ament o de Neurologia, FCM /UNICAM P - Caixa Post al 6111 - 13083-940 Campinas SP - Brasil. E-mail: [email protected]
M ULTI-M INICORE DISEASE REVISITED
Anamarli Nucci
1a, Luciano S. Queiroz
2a, Helder J.L. Zambelli
1b, José M art ins Filho
3cABSTRACT - Multi-minicore disease (MmD) is an infrequent congenital myopathy, defined by structural changes in opt ic and elect ron microscopy, namely, mult iple small areas lacking oxidat ive enzyme act ivit y and f ocal disorganizat ion of cont ract ile prot eins involving at most a f ew sarcomeres. The classical f orm of t he dise-ase manif est s as more or less severe hypot onia and generalized w eakness w it h predominance in axial and proximal limb muscles. Clinical variant s also exist . Usually M mD is inherit ed as an aut osomal recessive t rait . Genet ic het erogeneit y is recognized and up t o now mut at ions in t he genes of RYR1 and SEPN1 have been det ect ed. We record t hree unrelat ed cases of M mD. Case 1, w it h t he classical benign f orm, w as f ollow ed-up for 15 years. Case 2, presenting pharyngolaryngeal involvement and severe delay of head control, improved gradually, unt il independent gait w as acquired at age of six years. A moderat e rest rict ion of daily lif e act iv-it ies remains. Case 3, of ant enat al-onset , w as expressed by art hrogryposis of hands, predominance of scapu-lar girdle def icit and a st able course af t er t en years on physiot herapy. All cases w ere select ed by t he charac-t erischarac-t ic morphological abnormalicharac-t ies in biceps brachii samples, including eleccharac-t ron microscopy. Emphasis is given t o case 2 due t o t ype 1 f iber unif ormit y and mild endomysial f ibrosis, posing a diff icult diff erent ial diagnosis w it h congenit al muscular dyst rophy w ere it not f or t he signif icant number of mult i-minicores.
KEY WORDS: congenit al myopat hy, mult i-minicore disease, phenot ype, hist ochemist ry, elect ron microscopy.
M iopat ia dos m ult i-m inif ocos revisit ada
RESUMO - A miopatia dos múltiplos minifocos (MM) é doença congênita rara, definida por alterações estrutu-rais observadas ao microscópio ópt ico e elet rônico: múlt iplas e pequenas áreas sem at ividade enzimát ica oxidat iva e desorganização f ocal das prot eínas cont rát eis envolvendo poucos sarcômeros. A f orma clássi-ca da doença se manif est a com hipot onia mais ou menos grave e f raqueza generalizada, predominant e em músculos axiais e proximais em membros. Entretanto, variantes clínicas existem. A MM é usualmente herda-da como traço autossômico recessivo. Heterogeneiherda-dade genética tem sido reconheciherda-da e até o momento mu-t ações nos genes RYR1 e SEPN1 f oram demu-t ecmu-t adas. Relamu-t amos mu-t rês casos de M M . Caso 1, que mu-t em a f orma clássica e benigna da doença, assim permaneceu ao longo de 15 anos. Caso 2 apresent ou envolviment o f aringo-laríngeo e grave at raso no cont role cef álico que melhorou gradualment e, at é que a deambulação plena f oi adquirida aos seis anos; permanece com moderada limit ação das at ividades da vida diária. Caso 3 t eve início pré-nat al, expresso at ravés de art rogripose das mãos. Havia predominância de déf icit em cint u-ra escapular e o curso t em sido est ável, com f isiot eu-rapia, por 10 anos. Os casos f ou-ram selecionados pelas ca-ract eríst icas morf ológicas na biópsia do biceps braquial que incluiu microscopia elet rônica. Enf at izamos, no caso 2, a uniformidade das fibras do tipo 1 e a leve fibrose do endomísio, tendo sido necessário o diagnós-t ico dif erencial com disdiagnós-t rof ia muscular congênidiagnós-t a.
PALAVRAS-CHAVE: miopat ia congênit a, miopat ia dos mult i-minif ocos, f enót ipo, hist oquímica, microscopia elet rônica.
M ult i-minicore disease (M mD) is a rare
myopa-t hy def ined myopa-t hrough muscle biopsy (M B)
1. Cores are
well-demarcated, non membrane bound
mitochon-dria-f ree f oci in muscle f ibers, associat ed w it h
dis-organized myof ibrils. They may be single, cent ral
or eccent ric in t ransverse sect ions. In longit udinal
sect ions t hey st ret ch f or several sarcomeres or up
t o t he w hole lengt h of t he f iber, as classically seen
in cent ral core disease (CCD)
2,3, in w hich t hey
invol-ve almost alw ays t ype 1 f ibers. When mult iple
(de-signat ed mult icores by Engel et al.
4) and small
(na-med minicores by Ricoy et al.
5), cores may af f ect
A “ congenit al nonprogressive myopat hy w it h
mult if ocal degenerat ion of muscle f ibers” w as
brie-fly described in 1966 and designated multicore
myo-pat hy in 1971 by Engel et al.
4. Since t hen, at least
70 cases w ere published
1. The largest mult i-inst it
u-t ional series presenu-t ed 38 pau-t ienu-t s, leading u-t o
iden-tification of four phenotypically homogeneous
sub-groups
6.
MmD is transmitted as a recessive trait, although
sporadic cases are more f requent in pract ice
7.
Re-cently, studies provided evidence for genetic
hetero-geneit y of M mD
8-11. M ut at ion in t he skelet al
mus-cle ryanodine receptor (RYR-1) gene (locus 19q13)
8,9,11,
w ell recognized in CCD, has been f ound in cases
of MmD, thus linking the two diseases at least at t he
genet ic level. On t he ot her hand, classical cases of
MmD harbor a mutation in the selenoprotein N
(SE-PN-1) gene (locus 1p36)
10, w hich may also be f ound
in congenit al muscular dyst rophy w it h rigid spine
syndrome.
We describe t hree new sporadic cases of M mD
w it h dist inct clinical phenot ypes and course, f
ollo-w ed-up f or 10 t o 15 years. Recent insight s about
M mD are also discussed.
M ETHOD
Cases w ere select ed among t hose ref erred t o UNI-CAM P Neuromuscular Clinic f rom 1982 t o 2000 and w hich f ulf illed t he f ollow ing crit eria: 1. clinical evidence of congenit al myopat hy; 2. creat ine kinase (CK) analy-sis perf ormed at least once; 3. normal sensory and mot or nerve conduct ion velocit ies (NCV); 4. elect romyograph-ic examination (EMG); 5. MB showing mini-multifocal are-as devoid of oxidat ive enzyme act ivit y in a signif icant number of muscle f ibers and short lengt h disorganiza-t ion of myof ibrils observed uldisorganiza-t rasdisorganiza-t rucdisorganiza-t urally.
M B specimens (lef t biceps brachii) w ere f rozen in n-hexane previously cooled in liquid nit rogen and sect io-ned at 4 t o 8 µm in a cryost at . Serial sect ions w ere st ai-ned w it h hemat oxylin and eosin (H&E), modif ied Gomori t richrome (TRI), oil red O (ORO). Hist ochemist ry f or myo-f ibrillar adenosine t riphosphat ase (ATPase), reduced ni-cotinamide adenine dinucleotide dehydrogenase (NADH-TR) and succinat e dehydrogenase (SDH) w as perf ormed. A muscle sample w as f ixed in cacodylat e-buf f ered glu-t araldehyde, posglu-t f ixed in osmium glu-t eglu-t roxide, dehydraglu-t ed in et hanol and embedded in Araldit e®. Transverse and
longit udinal sect ions 1 µm t hick w ere st ained w it h t olui-dine blue. Ult rat hin sect ions w ere analyzed in a Zeiss-10 elect ron microscope.
CASES
Case 1. A male child w as born in Sept ember 1981 by
cesarean sect ion due t o pelvic present at ion. The parent s w ere healt hy and non consanguineous and denied
neu-romuscular disease in t heir f amilies. Floppiness w as de-t ecde-t ed early and a mild delay in mode-t or milesde-t ones w as observed by his pediat rician. In cont rast w it h good psy-chological development , running w as accompanied by f requent f alls. He had t o help himself w it h his hands in order t o st and up or climb st eps. When examined at 5 years and 10 months he was smart and collaborative, with moderat e dif f use hypot onia, f lat f eet and high arched palat e. M uscles w ere slender. Cervical f lexion and scapu-lo-humeral muscle f orces w ere graded 4+. Pelvic girdle def icit w as evident by Gow ers sign and a slight ly clum-sy gait . M uscle st ret ch ref lexes w ere present . No f acial muscle def icit , opht halmoparesis or ot her neurological abnormal f unct ion w ere det ect ed. NCV examined in up-per and lower limbs were normal as well as EMG. A serum CK sample revealed t w ice t he upper normal limit . Ano-t her sample, Ano-t aken a year earlier, had been normal. M B w as done at t he age of 6 years w hen t he diagnosis of M mD w as conf irmed. Hydrot herapy and physiot herapy w ere recommended and w eight gain w as discouraged. Prognat hism w as det ect ed at age 12 and correct ed by conservat ive ort hodont ic met hods, remaining mild. At age 19 and height of 1.90 m t he same mot or disabilit ies w ere observed. No signif icant vert ebral column abnorm-salit y developed, but a mild t horacolumbar scoliosis, sli-ght f lat t horax and mild bilat eral elbow cont ract ures w e-re seen.
Case 2. A f emale neonat e w as normally delivered in
September 1988 after a term gestation that was remarka-ble f or poor f et al movement s. She w eighed 2,460 g and measured 50 cm; head circumf erence w as 38 cm. She w as t he f irst child of young, healt hy and non consanguineous parent s t hat inf ormed t o be unaw are of muscle disease in t heir f amilies. Her younger sist er is healt hy. Severe hy-pot onia, w eak cry and bilat eral clubf oot w ere not ed at birt h. Despit e dif f icult y f or breast f eeding, t he child gradually acquired weight and height over the 50 percen-tile and could stay seated at the age of 15 months. Neuro-muscular consult at ion at t w o years and six mont hs w as mot ivat ed by poor head cont rol and marked delay in mot or milest ones, including inabilit y t o w alk. Physical examinat ion disclosed generalized hypot onia, dif f use amyot rophy, absent muscle st ret ch ref lexes and muscle w eakness (proximal exceeding dist al). Prominent cervi-cal def icit led t o abnormal head post ure. High arched palat e, elongat ed hands and bilat eral clubf oot w ere not ed. CK samples and NCV w ere normal. EM G of right
biceps brachii disclosed 100% of short durat ion
She w alked w it h aid at about 5 years of age and aut ono-mously at 6. Routine exams, EKG, thorax radiographs and cranial CT scans w ere non cont ribut ory. A moderat e rest rict ion of mot or perf ormance persist s.
Case 3. A male child was born in September 1981 after
normal pregnancy and cesarean delivery w eighing 3,120 g and measuring 52 cm. Parent s inf ormed general good healt h, non consanguinit y and absence of know n
neu-romuscular problems in t heir f amilies. Since birt h, dig-it s of bot h hands w ere kept in permanent f lexion and he had great dif f icult y in opening t hem. He w alked at about 18 mont hs and had f requent f alls specially w hen running. Cerebral palsy w as suspect ed, but his mot her ref used t o accept it . He w as able t o f ollow school t hough hampered by his hand def ormit y. Only physical t hera-py w as recommended. He w as f irst seen at our service in M arch 1993. On examinat ion, t here w as mot or def i-ciency of shoulder girdle w it h w inging of scapulae and dif f icult y in digit al ext ension. A mildly clumsy gait and slight general hypot onia w ere det ect ed. M uscle st ret ch ref lexes w ere normal. NCV and t w o CK samples proved normal and EM G w as myopat hic. M B w as done at age 11 leading t o diagnosis of M mD. The condit ion has kept st able up t o present [Fig 2 a-c].
DISCUSSION
MmD usually presents as congenital myopathy
2-7rarely as adult -onset disease
12. As regards t he
Bra-zilian lit erat ure, Werneck’s
13series of 1500 M B
in-cluded 47 cases of congenit al neuromuscular
disor-ders, of w hich f our w ere M mD. Anot her
congen-it al case w as published by Tsanaclis and Levi
14. Our
t hree cases are congenit al, t hough t hey w ere f irst
examined by us at 5 years and 10 mont hs, 3 years
and 6 mont hs and 11 years and 6 mont hs,
res-pect ively. M B w as perf ormed short ly t hereaf t er.
Our pat ient s have had a long f ollow -up t hat
al-low ed us t o recognize t he benign nat ural hist ory
of t he disease, as have ot hers
2,4,5. In spit e of slow
progression in most inst ances, f at al cases of M mD
have been described, generally associat ed w it h
car-diomyopat hy
15. Worse prognosis has also been
as-sociat ed w it h respirat ory insuff iciency due t o w
ea-kness of diaphragm and accessory respirat ory
mus-cles and/or t horacic def ormit ies
6. Respirat ory f
ailu-re occurailu-red in half of t he pat ient s of Jungblut h et
al.
7aged over 10 years and correlat ed w it h t he
de-gree of scoliosis.
Clinical manif est at ions vary great ly in M mD.
Ferreiro et al.
6could ident if y f our clinical phenot
y-pes: 1. Classical; 2. Severe f orm w it h
pharyngola-ryngeal involvement and lack of head cont rol; 3.
Ant enat al-onset w it h art hrogryposis; 4. Progressive
f orm w it h hand amyot rophy. Our case 1 is an
exam-ple of classical and benign M mD. Case 2 seemed t o
f it int o t he severe f orm w it h pharyngolaryngeal
in-volvement and lack of head cont rol during t he f irst
t w o years of lif e, but marked improvement of
mo-t or f uncmo-t ion w as observed af mo-t erw ards. Ambulamo-t ion
occurred lat e resembling t he cases of Hef f ner et
al.
16and Penegyres and Kakulas
17. Case 3 had ant
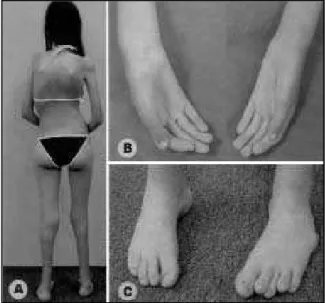
e-Fig 1. Case 2. Recent pict ure show ing dif f use muscle hypot
ro-phy and kyphoscoliosis (a); abnormal hands(b) and f eet (c).
Fig 2. Case 3. Winging of scapulae and w ast ing of shoulder gir-dle (a and b). In contrast, note good trophic appearance of pelvic
girdle and congenit al def ormit y of hands w it h f ailure t o
nat al-onset manif est ed by mild art hrogryposis of
hands. Weakness predominated in the shoulder
gir-dle, similar t hat observed by Jungblut h et al.
7in
f ive of t heir 19 cases. The myopat hy had a benign
and st able course.
Engel et al.
4described mild pt osis in one of t heir
t w o pat ient s. Anot her ocular f eat ure of t he disease
is ext ernal opht halmoplegia
18-20not ed in t he t w o
most severely affected patients of Jungbluth et al’s.
7series of 19. A challenging case of epilepsy, complex
encephalopat hy and minicore myopat hy w as
re-corded by Avoni et al.
21. M ult i-minicores have been
seen also in M arf an’s syndrome
22, short chain
acyl-CoA dehydrogenase deficiency
20, type III
glycogeno-sis
23and anhidrot ic ect odermal dysplasia
24.
Our t hree cases w ere select ed by t he charact
er-ist ic opt ic and elect ron microscopic abnormalit ies
on M B, as recommended
1. In case 1 a rout ine H&E
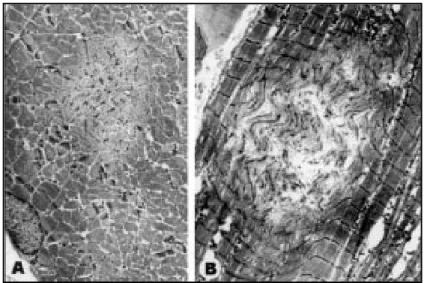
Fig 3. (a) Case 1. H&E. Variat ion in f iber size, int ernal nuclei, irregular basophilic areas, mult iple in some f ibers. (b) Case 1.
NADH-TR. M ult iple and variably sized f oci of decreased enzyme act ivit y. (c) Case 2. ATPase pH 4.3. Type 1 f iber unif ormit y. M any f ibers
show f oci of decreased or absent act ivit y w hich vary in size and number.
Fig 4. (a) Case 1. Elect ronmicrograph of t ransversely sect ioned muscle f iber. Small area of disorganizat ion of myof ibrillar st ruct ure w it h Z line f ragment at ion and
absence of mit ochondria (minicore). (b) Case 2. Similar st ruct ure in a longit
show ed slight variat ion of f iber diamet er,
numer-ous f ibers w it h cent ral nuclei, almost all exhibit ing
small and mult iple basophilic areas (Fig 3 a). Wit h
myof ibrillar ATPase bot h f iber t ypes w ere present
in similar numbers and bot h had mult i-minicores.
These f oci w ere negat ive f or oxidat ive enzymes
[NADH-TR (Fig 3 b) and SDH]. Type 1 f iber unif
or-mit y w as a peculiarit y of case 2 (Fig 3 c) and a
mo-derat e t ype 1 predominance w as seen in case 3.
Cases 2 and 3 also had signif icant numbers of
cen-t ral nuclei and mulcen-t i-minicores. In case 2 some
res-t ricres-t ed areas had mild perimysial and endomysial
f ibrosis and a more prominent variat ion in f iber
size, leading init ially t o t he diagnosis of
congeni-t al muscular dyscongeni-t rophy. Eleccongeni-t ron microscopy (Fig 4
a and b) and re-evaluat ion of t he f rozen sect ions
defined the diagnosis of MmD. This case was exclued
f rom our series of congenit al muscle dyst rophy
patients
25. We are thus in accordance with the
guide-lines of M mD consort ium
1t hat recommended
elec-t ron microscopy as one of elec-t he crielec-t eria f or
diagno-sis. Immunohist ochemist ry may help by
demons-t rademons-t ing f ilamin C and alpha-B-crysdemons-t allin w idemons-t hin demons-t he
minicores, where they react stronger than desmin
26.
Clinical genet ics and DNA st udies have show n
aut osomal recessive inherit ance in M mD. Rare
aut osomal dominant cases diagnosed as M mD
27,28have not yet been submit t ed t o genet ic
molecu-lar analysis. Molecumolecu-lar heterogeneity was conf irmed
because mut at ions in RYR1 as w ell as SEPN1 genes
have been ident if ied.
Apart f rom an anecdot al case w it h react ion t o
anest hesia
29no cases of malignant hypert hermia
have been document ed in M mD. M ore t han half
of the patients of Jungbluth et al.
7have been under
general anest hesia unevent f ully at least once. In
Ferreiro et al.
6series of 38 cases, 19 surgical
pro-cedures w it h general anest hesia w ere perf ormed
in 16 patients, with no abnormal reactions or
malig-nant hypert hermia being recorded in pat ient s or
t heir relat ives. In our pat ient s, M B (cases 1 and 2)
and cerebral CT scan (case 2) were performed under
propof ol or isof lurane w it hout problems. How ever,
considering that multi-minicores may be a transient
phenot ype of CCD
9, suscept ibilit y of pat ient s w it h
t his alt erat ion t o malignant hypert hermia should
alw ays be kept in mind. Theref ore, a caref ul
mon-it oring of t he pat ient by t he anest hesiologist and
ready availabilit y of dant rolene in t he operat ive
room are mandat ory.
Acknow ledgem ent s - We express our grat it ude t o
M ichael Davit t (Unit f or Prost hesis and Ort hosis -
UNI-CAM P) due t o skilf ul t echnical assist ance t o our pat ient (case 2).
REFERENCES
1. Ferreiro A, Fardeau M. 80thENMC international workshop on
multi-minicore disease; 1stinternational MmD workshop. Workshop report.
Neuromusc Disord 2002;12:60-68.
2. Shy GM, Magee KR. A new congenital non-progressive myopathy. Brain 1956;79:610-621.
3. De Cauwer H, Heytens L, Martin J-J. Workshop report of the 89thENMC
international workshop: central core disease. Neuromusc Disord 2002; 12:588-595.
4. Engel AG, Gomez MR, Groover RV. A recently recognized congenital myopathy associated with multifocal degeneration of muscle fibers. Mayo Clin Proc 1971;46:666-681.
5. Ricoy JR, Cabello A, Goizueta G. Myopathy with multiple minicore: report of two siblings. J Neurol Sci 1980;48:81-92.
6. Ferreiro A, Estournet B, Chateau D, et al. Multiminicore disease -Searching for boundaries: phenotype analysis of 38 cases. Ann Neurol 2000; 48:745-757.
7. Jungbluth H, Sewry C, Brown S C, et al. Minicore myopathy in chil-dren: a clinical and histopathological study of 19 cases. Neuromusc Disord 2000;10:264-273.
8. Jungbluth H, Müller CR, Halliger-Keller B, et al.Autosomal recessive
inheritance of RYR1 mutations in a congenital myopathy with cores. Neurology 2002;59:284-287.
9. Ferreiro A, Monnier N, Romero N B, et al. A recessive form of central core disease, transiently presenting as multi-minicore disease, is asso-ciated with a homozygous mutation in the ryanodine receptor type 1 gene. Ann Neurol 2002;51:750-759.
10. Ferreiro A, Quijano-Roy S, Pichereau C, et al. Mutations of the selenopro-tein N gene, with is implicated in rigid spine muscular dystrophy, cau-se the classical phenotype of multiminicore dicau-seacau-se: reascau-sessing the nosology of early-onset myopathies. Am J Hum Genet 2002:71:739-749. 11. Monnier N, Ferreiro A, Marty I, Labarre-Vila A, Mezin P, Lunardi J. A homozygous splicing mutation causing a depletion of skeletal muscle RYR1 is associated with multi-minicore disease congenital myopathy with ophthalmoplegia. Hum Mol Genet 2003;12:1171-1178. 12. Bonnette H, Roelofs R, Olson W H. Multicore disease: report of a case
with onset in middle age. Neurology 1974;24:1039-1044.
13. Werneck LC. Correlação entre a incapacidade funcional, idade e enzi-mas séricas nas doenças neuromusculares. Arq Neuropsiquiatr 1995;53:60-68.
14. Tsanaclis AMC, Levy JA. Doença do “minicore”: relato de um caso. Rev Hosp Clin Fac Med S Paulo 1982;37:241-243.
15. Shuaib A, Martin JME, Mitchell LB, Brownell AKW. Multicore myopa-thy: not always a benign entity. Can J Neurol Sci 1988;15:10-14. 16. Heffner R, Cohen M, Duffner P, Daigler G. Multicore disease in twins.
J Neurol Neurosurg Psychiatry 1976;39:602-606.
17. Penegyres P K, Kakulas B A. The natural history of minicore-multicore myopathy. Muscle Nerve 1991;14:411-415.
18. Swash M, Schwartz MS. Familial multicore disease with focal loss of cross-strations and ophthalmoplegia. J Neurol Sci 1981;52:1-10. 19. Gordon PH, Hays AP, Rowland LP, et al. Erroneous diagnosis
correct-ed after 28 years: not spinal muscular atrophy with ophthalmoplegia but minicore myopathy. Arch Neurol 1996;53:1194-1196.
20. Tein I, Haslam RHA, Rhead WJ, Bennett MJ, Becker LE, Vockley J. Short-chain acyl-CoA dehydrogenase deficiency: a cause of ophthal-moplegia and multicore myopathy. Neurology 1999:52:366-372. 21. Avoni P, Monari L, Carelli V, et al. Congenital encephalomyopathy
with epilepsy, chorioretinitis, basal ganglia involvement, and muscle minicores. Ann Neurol 2000;47:395-399.
22. Pagès M, Echenne B, Pagès AM, Dimeglio A, Sires A. Multicore disease and Marfan’s syndrome: a case report. Eur Neurol 1985;24:170-175. 23. Pellissier JF, Barsy T, Faugere MC, et al. Type III glycogenosis with
mul-ticore structures. Muscle Nerve 1979;2:124-132.
24. Gordon CP, Litz S. Multicore myopathy in a patient with anhidrotic ecto-dermal dysplasia. Can J Anaesth 1992;39:966-968.
25. Nucci A. Distrofia muscular congênita: variações fenotípicas em 27 casos. Tese, Universidade Estadual de Campinas. Campinas, 1992. 26. Bönnemann CG, Thompson TG, van der Ven PFM, et al.Filamin C
accu-mulation is a strong but nonspecific immunohistochemical marker of core formation in muscle. J Neurol Sci 2003;206:71-78.
27. Vanneste JA, Stam FC. Autosomal dominant multicore disease. J Neurol Neurosurg Psychiatry 1982;45:360-365.
28. Paljärvi L, Kalimo H, Lang H, Savontaus M-L, Sonninen V. Minicore myopathy with dominant inheritance. J Neurol Sci 1987;77:11-22. 29. Koch BM, Bertorini TE, Eng GD, Boehm R. Severe multicore disease