Review
0103 - 5053 $6.00+0.00*e-mail: [email protected]
Overview of Ligand
versus
Metal Centered Redox Reactions in Tetraaza Macrocyclic
Complexes of Nickel with a Focus on Electron Paramagnetic Resonance Studies
Joshua Telser*
Department of Biological, Chemical and Physical Sciences, Roosevelt University, 430 South Michigan Avenue, Chicago, 60605-1394 IL USA
Complexos de cobre(II) (3d9, S = 1/2) são estáveis e amplamente investigados por espectroscopia
de ressonância paramagnética eletrônica (EPR). Já o isoeletrônico níquel(I) é muito menos comum e muito menos estudado. No entanto, níquel(I) tem interesse biológico, uma vez que o sítio ativo da metil coenzima M redutase (MCR) contém um ligante macrocíclico, F430, que coordena o NiI
na sua forma ativa, MCRred1. Assim, o comportamento redox e espectroscópico de complexos tetraazamacrocíclicos de níquel tem importância na química biomimética. O estudo desses complexos é complicado pela diiculdade na obtenção de NiI a partir dos precursores estáveis de NiII. A redução
de complexos macrocíclicos de NiII pode gerar NiI em certos casos, mas em muitos outros leva à
redução do macrociclo, gerando um ânion radical orgânico. Estudos anteriores da formação de complexos tetraazamacrocíclicos de NiI são aqui discutidos em termos da competição entre a redução
centrada no metal e a centrada no ligante. Resultados de EPR são particularmente importantes para distinguir esses dois processos de redução, já que a formação de NiI produz espectros de EPR
característicos, similares aos de CuII, enquanto a redução centrada no ligante gera espectros de EPR
agudos, centrados em g = 2,00 e típicos de radicais orgânicos. Mesmo que uma redução centrada no metal ocorra, a geometria do complexo macrocíclico de NiI resultante é amplanente variável e,
consequentemente, o espectro de EPR também será. Nesse caso, a comparação é entre os extremos dos espectros típicos de complexos tetragonais distorcidos (estado fundamentaldx2–y21, que inclui as
geometrias octaédrica tetragonalmente distorcida, piramidal de base quadrada e quadrado-planar) e dos complexos bipiramidais de base trigonal (estado fundamentaldz21). Trabalhos anteriores
realizados com CuII foram relacionados com a situação para NiI. Os diferentes tipos de espectros de
EPR desses sistemas são discutidos especiicamente usando exemplos inéditos de vários complexos tatraazamacrocíclicos de níquel, incluindo F430 e a própria MCR.
Copper(II) (3d9, S = 1/2) complexes are stable and widely investigated by electron paramagnetic
resonance (EPR) spectroscopy. In contrast, isoelectronic nickel(I) is much less common and much less investigated. Nickel(I), however, is of biological interest as the active site of methyl coenzyme M reductase (MCR) contains a tetraaza macrocyclic ligand, F430, which coordinates NiI in its active
form, MCRred1. As result, the redox behavior and spectroscopy of tetraaza macrocyclic complexes of nickel is of importance in biomimetic chemistry. Such efforts are complicated by the dificulty in generating NiI from their stable, NiII, precursors. Reduction of NiII macrocyclic complexes can
afford NiI in certain cases, but in many other cases can lead instead to reduction of the macrocycle
to generate an organic radical anion. Previous studies on the formation of tetraaza macrocyclic complexes of NiI are discussed in terms of the competition between metal-centered and
ligand-centered reduction. EPR results are particularly important in making the distinction between these two reduction processes, as formation of NiI gives characteristic EPR spectra similar to those for CuII,
while ligand-centered reduction gives narrow EPR spectra at g = 2.00, typical of organic radicals.
Even if metal-centered reduction occurs, the geometry of the resulting NiI macrocyclic complex
is highly variable and, as a result, the EPR spectral appearance is highly variable. In this case, the comparison is between the extremes of spectra typical for tetragonally distorted complexes (dx2–y21
ground state, which includes tetragonally distorted octahedral, square pyramidal and square planar geometries) and those for trigonal bipyramidal complexes (dz21 ground state). Previous work on CuII
was related to the situation for NiI. The different types of EPR spectra for such systems are speciically
discussed using previously unpublished examples of several tetraaza macrocyclic complexes of nickel, including F430 and MCR itself.
the EPR spectra of a AgII porphyrin can be analyzed
analogously to the corresponding CuII complex.18 Other
possibilities lie outside of Group 11. These could include Group 9 complexes in the zero oxidation state, e.g., Co0;
however, such species are more realistically considered as organometallic radicals and are typically found in di- or polynuclear complexes, such as diamagnetic [Co2(CO)8].19
The most viable candidate is in Group 10, namely NiI.
Relative to NiII, NiI is uncommon; however, pioneering
work by Busch and co-workers20 has shown the accessibility
of a variety of coordination complexes of NiI. At that
time, NiI complexes were of interest only to coordination
chemists; however, the discovery soon thereafter of the enzyme methyl CoM reductase (MCR) changed that situation dramatically.21-23 MCR catalyzes the inal step in
methane generation by archaea, a process by which most of biogenic methane is created.22,24-26 MCR is found in several
microorganisms, of which that from Methanothermobacter
marburgensis is the best characterized (the taxonomy of
these organisms is complicated and has been changed over the years; older papers on MCR refer to this organism as
Methanobacterium thermoautotrophicum strain Marburg).
MCR contains at the active site a prosthetic group comprising a unique macrocyclic ligand, known as F430 (based on its maximum absorption wavelength), a diagram of which is shown below.27,28 In contrast to tetrapyrroles,
F430 is a monoanion and is much more saturated. Each pyrroline ring has signiicantly different substituents and is identiied by the letters A through D, so that the upper left ring in the diagram below is denoted A, the upper right (with lactam substituent) is B, the lower right is C, and the lower left (with cyclohexanone substituent) is D. F430 is relatively thermally unstable and can epimerize to give the 12,13-diepimer of the propionic acid side chains on ring C; shown below with the ring designations.28,29
In the resting state, inactive enzyme, F430 contains a NiII
ion, which is EPR silent at X-band, but has been studied by magnetic circular dichroism (MCD).30,31 However, the
active form, MCRred1, contains NiI,32-35 as does a related
form, MCRred2.36 X-ray crystallography has been possible
on the relatively stable, NiII forms of MCR,37,38 but not
on the reactive, NiI forms. The crystal structure of the
pentamethylester of F430, F430M, has also been reported (as the 12,13-diepimer, since this is the thermally stable form; CSD code: KOBCEJ).39
1.3. Model complexes for MCR that are porphyrin-derived
The discovery of MCR led to a reawakening of interest in the coordination chemistry of NiI and speciically in
model chemistry of MCRred1. Synthesis of the full structure
1. Introduction
1.1. General background on electron paramagnetic resonance (EPR) spectroscopy
Electron paramagnetic resonance (EPR) spectroscopy has been widely applied over the past six decades to the study of coordination complexes of the d block (transition metal) ions.1,2 Among the many possible dn electronic conigurations found, the d9 coniguration has been
particularly well studied.1-6 This is the case for several
reasons, chemical and physical. In the chemical context, the d9 coniguration is best represented by CuII, which
forms a vast number of stable coordination complexes,7
many of which have biological relevance.3,8-10 In the
physical context, the d9 (S = 1/2) coniguration is very
amenable to study by EPR spectroscopy since there are no complications from intermolecular electron-electron interactions in mononuclear complexes. As long as the CuII sites are suficiently diluted, there are
no intramolecular electron-electron interactions either, although these can be observed in undiluted solids.11
It should also be noted that EPR spectra of multi-CuII
centers can be intricate due to intramolecular exchange coupling.12 Equally important, the EPR spectra of d9
systems are highly informative in terms of providing information on molecular geometry and chemical bonding. This utility was demonstrated many years ago for CuII coordination complexes by Maki and
McGarvey,13,14 and a more qualitative analysis of CuII
EPR spectra has been very useful in bioinorganic chemistry.3 In contrast, mononuclear complexes with
multiple electron/holes, however, such as those with the d8 electronic coniguration (NiII in many coordination
environments, such as tetrahedral and octahedral), often exhibit complicated intramolecular electron-electron interactions that arise from spin-orbit and spin-spin coupling.1,15 These effects can lead to signiicant
zero-ield splitting (zfs) and hence dificulty in obtaining EPR spectra at conventional microwave frequencies (i.e.,
X-band: ca. 9 GHz). Use of high frequencies (> 95 GHz)
combined with high magnetic fields (up to 25 T), however, can yield EPR spectra of such “EPR-silent” NiII
complexes, both four-coordinate16 and six-coordinate.17
1.2. Background on nickel(I) and on methyl CoM reductase (MCR)
Other than CuII, what transition metal ions have the
d9 electronic coniguration? Silver(II) is uncommon, but
of the F430 cofactor would be a daunting task; however, the salient features of the electronic structure of the NiI
ion can be reproduced by much simpler complexes. These include some of the relatively more saturated tetraaza macrocyclic complexes irst reported by Busch and co-workers,20 and of the relatively less saturated, porhyrinic
complexes described by Fajer and Stolzenberg and their co-workers.40-50 Among these models, the most fruitful has
been that of Ni with the ligand octaethyisobacteriochlorin (OEiBC), a diagram of which is shown below. The octaethyl substituents aid in solubility, but may have other electronic effects. The stereochemistry at the four saturated positions (reduced cis pyrrole (pyrroline) rings A (or C) and B (or
D), applying the F430 nomenclature to the diagram below) that distinguish OEiBC from its standard porphyrin
analog octaethylporphyrin (OEP) is not speciied. The bacteriochlorin (OEBC) has reduced trans pyrrole rings
(i.e., rings A/B and C/D), but has been much less studied
in terms of Ni chemistry. In between the porphyrin and iBC/BC in terms of saturation is the chlorin, in which only one pyrrole has been reduced,51 also shown below with
unspeciied stereochemistry.
There is also the “triply” reduced form, in which only one ring remains a pyrrole, known as octaethylpyrrocorphin (OEPC). The synthesis and crystal structure of [NiII(OEPC)]
have been reported,52 but, to our knowledge, no investigations
of its reduction chemistry have been reported.
Ni(OEiBC) is prepared in the NiII form (as are
[Ni(OEP)], [Ni(OEBC)], [Ni(OEC)]), but can be reduced electrochemically,47 or by Na(Hg) amalgam in dry
organic solvents to yield the NiI complex in solution,
[Ni(OEiBC)]−.53 Other NiI isobacteriochlorin (iBC)
complexes, which contain the fused cyclohexanone ring of F430, can be analogously prepared.44 [Ni(OEiBC)]− has not
only spectroscopic relevance to MCRred1, but also exhibits reactivity that has some similarities to that of MCR.42,48
What is striking about the effectiveness of OEiBC as a model ligand for F430 is how structurally different the two are. F430 is a much more highly saturated and more lexible macrocycle than OEiBC,54 although porphyrinic
macrocycles should not be thought of as the rigid disks by which they are so often depicted. Extensive studies by Ghosh and co-workers55,56 have probed the conformational
lexibility and deformations on porphyrinic complexes. Even more puzzling is that among the various NiII
porphyrinic complexes, only iBCs are successfully converted into NiI.44 The fully unsaturated, π-conjugated
OEP complex of NiII yields a ligand-centered radical
upon reduction,47 although for the chlorin analog, an EPR
spectrum of [NiI(OEC)]− can be transiently observed.49
A complication with these complexes when undergoing chemical reductions is formation of phlorins, in which
meso positions are reduced. Stable, square planar (sq pl),
N
N N
N
Ni
H
O
CO2H
CO2H CO2H
HN O
H HO2C
H2NOC
HO2C
F
430N
CO2H CO2H 12
13
12,13-diepimer
C
A
B
D
N
N N
N
Ni
Ni(OEiBC)
H
H H
H
N
N N
N
Ni
Ni(OEBC)
N
N N
N
Ni
Ni(OEC)
H
H H
H
diamagnetic NiII phlorins result eventually from reduction
of both [Ni(OEP)] and [Ni(OEC)].49 Concerning the
closer models to F430, namely those with the fused cyclohexanone ring, in both their porphyrin and chlorin forms (shown below), reduction gives stable complexes well characterized in solution by EPR, optical, and X-ray spectroscopic techniques. For both of these complexes, the EPR spectra exhibit a very slight g anisotropy indicating
a small contribution from spin density on Ni 3d orbitals, however these species can by no means be considered as authentic NiI.44
The ligand-centered reduction might be expected for the porphyrins (OEP and the F430 model), since they are as different from F430 as is possible in terms of π-conjugation and thus have the greatest availability of ligand-centered orbitals of suitable (low) energy to be electron acceptors. The fused cyclohexanone ring, while leading to a closer model for authentic F430, appears to have no effect at preventing ligand-centered reduction. Renner et al.44 also
prepared hexahydro- and octahydroporphyrins (structural diagrams shown below; note that there are two regioisomers of the hexahydroporphyrin (CSD code: KODHAM), depending on which one of the two meso alkenes is reduced;
both are reduced in the octahydroporphyrin shown on the right; the hydrogens added to the meso positions are not
shown). These tetraaza macrocycles are lessπ-conjugated than the iBCs and reproduce the structure of F430 as closely as one could reasonably hope for, yet they yield even more purely ligand centered (π-anion) radicals upon reduction, as shown by EPR spectra that consist of a narrow signal at
g = 2.0029 (essentially the free electron value, ge = 2.0023,
so that there are no d orbital contributions to the SOMO whatsoever).44
Although EPR spectroscopy is a convincing indicator of metal versus ligand-centered reduction, Renner et al.44 also
employed X-ray absorption spectroscopic methods (XAS, EXAFS) that independently show the reduction of NiII to
NiI and the associated changes in Ni-N bond lengths. The
larger NiI ion can be accommodated by a distortion in which
two Ni-N bonds lengthen signiicantly, while the other two shorten slightly relative to the NiII parent complex. Thus
the ability of the speciic macrocycle to adjust to the size changes in the nickel ion contributes what is in a sense a steric effect in determining the site of reduction.47
1.4. Model complexes for MCR that are saturated macrocycle-derived
If one then begins from the other direction, namely the totally saturated macrocycle 1,4,8,11-tetraazacyclo-tetradecane ([14]aneN4, cyclam), and its variously methyl substituted analogs (speciically, Me6[14]aneN4, shown below), then NiI complexes result upon electrochemical
reduction of the NiII parent complex.20 This result is
perhaps the only one that is readily expected since there are no ligand-based π MOs to act as electron acceptors. However, introduction of only minimal π-bonding into the macrocyclic ligand can lead to generation of ligand-centered, as opposed to metal-centered (i.e., NiI) reduction
products. The results are summarized in the scheme below, where “NiI” indicates metal-centered reduction (upper row
of diagram) and “• −” indicates ligand-centered reduction
(lower row). Lovecchio, Gore and Busch studied a number of other such complexes, however the scheme below depicts the salient macrocyclic ligand types.20 Related
studies were subsequently performed by Gagné and co-workers57 on these and analogous complexes with borate
linked bisdimine ligands (not shown). A number of these complexes were later studied by EXAFS by Furenlid et al.58
In this wide range of macrocyclic complexes, as long as the imino groups are fully π-isolated, then the reduction is metal centered; all that is necessary for ligand-centered reduction is to have a single conjugated α-diimine functionality.57
Complexes of NiII with acyclic, as opposed to macrocyclic,
α-diimine ligands ([(R'N=C(R)C(R)=NR')MX2];
M = NiII, PdII; X = halide, alkyl etc) are of great interest in
their own right, due to their activity as alkene polymerization catalysts.59,60 It should also be noted that the related,
β-diketiminate ligand (NacNac, (RC(=NR′)CH(=NR′)CR)−)
has been widely used for a wide variety of d and p block metal ions, and many of these complexes have catalytic
N
N N N
CO2Me
Ni
O
N
N N N
H
CO2Me
Ni
H
O
F430 model porphyrin F430 model chlorin
N
N N N
H
CO2Me Ni
H H
H
O H
N
N N N
H
CO2Me Ni
H H
H
O H H
F430 model hexahydroporphyrin F430 model octahydroporphyrin ~
~ ~
activity as well.60,61 Bai et al.62 provide an example of such
a NiII complex, and also provide a comprehensive listing
of references on β-diketiminato complexes. These workers have also isolated NiIβ-diketiminato complexes,63 which
indicates that the β-diketiminate ligand is not reduced, despite its extensive π-conjugation. A point that to my knowledge has not been made before is that F430 itself can be thought to contain a β-diketiminate group, as shown below in red, which is not the case for any of the [14]1,4,8,11-di- or tetraene complexes shown above.
1.5. Computational studies on tetraazamacrocyclic Ni complexes
There are no obvious “rules of thumb” for a simple coordination chemist to use as guidelines as to whether a given NiII complex with amino/imino ligands will be reduced
to a NiI complex, or to an organic radical anion species. Only
the extrema in terms of macrocyclic ligand π-conjugation can be easily deined in that no π-conjugation (e.g., fully
saturated [14]aneN4) gives NiI, and maximum π-conjugation
(e.g., fully unsaturated (aromatic) OEP) gives a
ligand-centered radical. This problem thus represents a potentially fruitful area for application of computational methods, and indeed such studies have already been performed on MCR/F430 and related macrocyclic model systems.64-69
Of particular relevance is the very recent study by Ryeng, Gonzalez and Ghosh.69 These workers performed
an extensive DFT study of a carefully selected series of
Ni hydroporphyrin complexes. These included chlorin, iBC, and BC ligands with no substituents, and each with tetramethyl and octaethyl substituents. Complexes with heteroatom substitution, i.e., oxa- and thiaporphyrins, which
have been studied experimentally,70 were also investigated
computationally; however, these are not relevant to the present discussion which is limited to tetraaza complexes of Ni. As is characteristic of the Ghosh group, the results are very comprehensive. We point out here only that relative to the NiII parent complex, [NiI(OEC)]− and [NiI(OEBC)]−
are actually calculated to be at lower energy than their ligand-reduced forms (by 0.2-0.3 eV). Apparently, these forms are not suficiently stabilized in solution to persist indeinitely, suffering from other reaction pathways, such as phlorin formation. The calculation for [NiI(OEiBC)]−,
however, indicates that this form is much lower in energy (by 0.55 eV) than the ligand-reduced form, [NiII(OEiBC•)]−,
which apparently leads to its stability in solution. This stability has allowed the full EPR/ENDOR spectroscopic characterization of [NiI(OEiBC)]−.50 This energetic result
is more the consequence of relative instability of the
ligand radical anion than of relative stability of the NiI
form. Conformational lexibility in the OEiBC macrocycle relative to the more rigid OEC and OEBC (and presumably OEP) is the crucial factor in stabilizing the NiI form. This
quantitative result from computations agrees with earlier, qualitative proposals.49,51
Wondimagegn and Ghosh68 had earlier studied F 430 itself
and shown that this unique ligand has unique conformational characteristics that help support the NiI species observed
by a variety of spectroscopic methods.40,41,50 Nevertheless,
the situation with more reduced, and presumably more lexible, macrocyclic complexes, such as those studied by the groups of Busch,20 and Gagné,57 has yet to be resolved.
2. EPR Results for Tetraazamacrocyclic Ni
Complexes
2.1. Overview of case studies of individual Ni complexes
We describe here EPR studies on several macrocyclic complexes of nickel that span a variety of tetraaza macrocycle coordination. Also included are EPR spectra of the isolated MCR cofactor, F430, in its reduced, NiI form
(NiIF
430), together with the holoenzyme form that contains
this species, MCRred1. In the case of enzymes, introduction of magnetically active nuclei is often much more feasible than in model complexes. The anaerobic organisms that are the source of MCR can be grown on medium enriched in, e.g., 61Ni (I = 3/2, 1.13% natural abundance), whereas
chemical synthesis using such isotopes is very expensive. N
N N N
Ni
[NiI(Me 6[14]aneN4)]+
H H H H N N N N Ni H H N N N N Ni
[NiI(Me
6[14]4,11-dieneN4)]+ [NiI(Me6[14]1,4,8,11-tetraeneN4)]+
N
N N N
Ni
[NiII(Me
2[14]1,3-dieneN4) ]+
H H N N N N Ni
[NiII(Me
6[14]1,3,7,11-tetraeneN4) ]+[NiII(Me4[14]1,3,8,10-tetraeneN4) ]+
Such isotopologs deinitively showed the role of nickel in MCR.22,32,35
Concerning the tetraaza macrocyclic model complexes for MCR, we irst present the EPR spectra of
tct-[NiI(OEiBC)]−, which represents the most unsaturated
macrocycle to give a stable NiI species in solution; the
ttt- and tct- isomers (see diagram below) gave identical
EPR results. No solid NiI OEiBC complex has been
isolated. Surprisingly, to our knowledge, no crystal structure of [NiII(OEiBC)] has been reported (nor of
[Ni(OEC)]), although structures of [NiII(OEBC)] (CSD
code: DEGTAK52) and [NiII(OEP)] (several structures,
of which the most recent has CSD code: NOEPOR0271),
and [Ni(OEPC)] (CSD code: DEGSUD52) are known.
However, the crystal structures of the PdII series [Pd(OEP)],
[Pd(OEC)], and tct-[Pd(OEiBC)] have been reported by
Stolzenberg et al.72 The larger size of PdII allowed a better
probe of the effect of ring reduction than for the NiII analogs.
Lastly, the crystal structures of the series [Ni(TMP)], [Ni(TMC)] and [Ni(TMiBC)] (where TMiBC = dianion of 5,10,15,20-tetramethylisobacteriochlorin, and analogously for TMP and TMC) have been reported;73,74 however, these
tetrapyrroles have substituents unlike those of F430 (i.e., at
the meso positions, rather than at the β positions (pyrroles/ pyrrolines)) and are considered here only in passing. The relative stability of [NiI(TMC,TMBC,TMiBC)]−
versus [NiII(TMC,TMBC,TMiBC•)]− has been studied
computationally by Ryeng et al.,69 who showed that
ligand-centered reduction is energetically favored for the TMC and TMBC complexes, but is less favored (by ca.
0.55 eV; similar to the result for OEiBC) for the TMiBC complex. Despite this, we are not aware of any report of a NiI species upon reduction of Ni(TMiBC). Perhaps the
recent work of Ryeng et al. 69 will inspire a reinvestigation
of this process in the meso-substituted NiII tetrapyrrole
series.
Moving in the direction of greater saturation, we also describe studies on a nickel complex of a diene derivative of 1,4,8,1l-tetraazacyclotetradecane, Me6 [14]4,11-dieneN4 (formally 5,7,7,12,14,14-hexamethyl-1,4,8,11-tetraazacyclotetradeca-4,11-diene), in which there is no conjugation of the two imines, so that a NiI species is
formed upon reduction.20,57,58 Two geometrical forms of this
complex are found, rac and meso, as shown in the diagram
below, and each has been structurally characterized in the NiII state (CSD codes: KUGNEF (meso), MAZTNI02
(rac)).75 The structure of only the meso form has been
determined for NiI (as [Ni(Me
6[14]4,11-dieneN4)](ClO4),
CSD code: KINNOK).58 The speciic NiI solid state sample
studied here was a mixture of these rac and meso forms;
this heterogeneity is maintained in solution.
The final isolable tetraaza macrocyclic complex to be described is that of the fully saturated ligand 1,4,8,1l-tetramethyl-1,4,8,1l-tetraazacyclotetradecane (tetramethylcyclam, tmc, [14]ane(NMe)4),76 for which two
stereoisomers are available as in the diagrams shown below. Crystal structures of a variety of [NiII(tmc)]2+ complexes,
several with axial ligands, but none with nitrile(s), have been reported; that most relevant to this study is
RRSS-[NiII(tmc)](CF
3SO3)2 (CSD code: DONCAK),77
which is a rigorously sq pl complex. It must be noted that although isomerically pure [NiII(tmc)]2+ complexes
can be isolated, this isomeric integrity is not maintained upon reduction. Chemical reduction of either NiII pure
isomer yields solutions containing both the RSRS- and RRSS-[NiI(tmc)]+ isomers.76,78 For solubility reasons, the
RRSS isomer crystallized selectively, as RRSS-[NiI(tmc)]
(CF3SO3)•NaCF
3SO3 (CSD code: ZIMWUN),76 however
the solutions studied here contain both isomers, albeit in unknown proportion. At equilibrium in aqueous solution, the RRSS/RSRS ratio is roughly 3:1.78
Related studies by Meyerstein and co-workers79,80 on
a variant of tmc with macrocycle methylation (oficially,
N
N N N
Ni
tct-[NiI(OEiBC)]−
ttt-[NiI(OEiBC)]−
N N
Ni
N
N N
N
Ni H
H
[NiI(Me6[14]4,11-dieneN4)]+
N
N N
N
Ni
H
H
1RS,4RS,7RS,8SR,11SR,14SR
)-1,4,5,5,7,8,11,12,12,14-decamethyl-1,4,8,11-tetraazacyclotetradecane; referred to herein as C-meso-[Me6[14]ane(NMe)4], or as Me6tmc;
diagram shown below) also showed the stability of NiI.
The structure of only the NiII form of this complex has
been reported (as [NiII(Me
6tmc)](ClO4)2; CSD code:
DUKPUU).81
Lastly, we describe the EPR spectra of the unstable species formed upon γ-irradiation of both [NiII(OEiBC)]
and [NiII(OEP)] at 77 K. This cryoreduction technique, in
which γ-irradiation ejects electrons from the appropriate solvent (various organic solvents, such as ethanol, or water/ glycerol) has been pioneered by Davydov and applied to a wide variety of metalloproteins, including diiron-oxo proteins,82 iron-sulfur proteins,83 heme proteins,84,85 and
MCR itself.86 These new results show that it is possible
to generate a NiI porphyrin, but that it can survive only
at cryogenic temperatures. This work is analogous, but in striking contrast, to the studies using UV-irradiation in luid solution, followed by freezing in liquid nitrogen, which showed only the generation of an anion radical, [NiII(OEP•−)].49 In a related technique, radiolysis (pulsed
or steady-state), using electrons generated by a linear accelerator, has also been used to generate NiI from tetraaza
macrocyclic NiII complexes.79,80
2.2. Sources of complexes described in case studies and experimental protocol
The complexes studied were obtained from a variety of sources. Samples of Methanothermobacter marburgensis
MCRred1 were provided by Prof. Stephen W. Ragsdale (University of Michigan, Ann Arbor, MI, USA) and prepared by reduction with TiIII citrate as described previously.87
Isolated native F430 and its 12,13-diepimer were provided by Prof. Robert A. Scott (University of Georgia, Athens, GA, USA) and converted in his laboratory to their NiI forms
by reduction with TiIII citrate as described previously.50
RRSS-[NiI(tmc)](CF
3SO3)•NaCF3SO3 was provided by Prof.
Charles G. Riordan (University of Delaware, Newark, DE, USA) and prepared in his laboratory following literature procedures.76 [NiI(Me
6[14]4,11-dieneN4)](ClO4) (mixture
of rac and meso forms) was provided by Dr. Etsuko
Fujita, Brookhaven National Laboratory, Upton, NY, USA and prepared in her laboratory following literature procedures.57,58 The NiI forms of these complexes were
provided as solids and then dissolved under nitrogen atmosphere in dry n-butyronitrile/n-propionitrile (9:7 v/v),
which mixture forms a good glass for EPR spectroscopy. The complexes ttt- and tct-Ni(OEiBC) were prepared
and chromatographically separated by Dr. Mark W. Renner (Brookhaven National Laboratory) as described previously.50 The NiI forms of these complexes were
generated in Dr. Renner’s laboratory by reduction using Na(Hg) amalgam in dry 2-methyltetrahydrofuran (2-Methf) solution,50 and shipped at low temperature for EPR
measurements at Northwestern University. The complex [NiII(OEP)] was obtained from Porphyrin Products (now
Frontier Scientiic, Logan, UT, USA).
EPR spectra at 9.0-9.7 GHz (X-band) of MCRred1 samples were recorded by Dr. Yih-Chern Horng at the University of Nebraska, Lincoln, NE, USA on a Bruker ESP 300E spectrometer. EPR spectra at 34-36 GHz (Ka-band, often, but erroneously, referred to as Q-band) were recorded on a modified Varian spectrometer at Northwestern University, Evanston, IL, USA. Experimental conditions are given in the igure captions. The 35 GHz spectra were recorded under “passage” conditions,88 so that the signal appears as an absorption,
rather than irst derivative lineshape. The igures generally present digital derivatives in addition to or instead of the original, passage spectrum so that the appearance is consistent with typical EPR spectra, such as those reported elsewhere for such NiI species.
NiII(OEP) and NiII(OEiBC) samples in 2-Methf
solution were γ-irradiated at 77 K. The irradiation was done by Dr. Roman Davydov, Northwestern University, using a Gammacell 200 60Co irradiator at the University
of Chicago Pritzker School of Medicine, using procedures developed by him.82,84,89-91 The irradiated samples were
maintained at 77 K (or lower) throughout the subsequent EPR spectroscopic measurements.
N N
N N
Ni
RSRS-[NiI(tmc)]+ RRSS-[NiI(tmc)]+
N N
N N
Ni
R
R R
R
S S
S S
N
N N
N
Ni
C-meso-[NiI(Me6[14]ane(NMe)4)]+,
All computer programs for EPR simulation (QPOWA, written originally by Belford and co-workers at the U. of Illinois, Urbana, IL, USA,92,93 and DDPOWH) and ligand
field analysis (DSOXF, DDN package) are written in FORTRAN (g77) and are available from the author.
2.3. 35 GHz EPR Spectra of NiIF
430 and [Ni
I(OEiBC)]−
Among the various tetraaza macrocyclic complexes of NiI studied here, the simplest EPR spectrum is that
for the most structurally elaborate macrocycle, namely F430. Figure 1 presents 35 GHz EPR spectra of NiIF
430
and tct-[NiI(OEiBC)]−. The EPR parameters for these
and other NiI species are summarized in Table 1. Use of
higher microwave frequencies, here 35 GHz, often reveals rhombicity that is not resolved at X-band (ca. 9 GHz). This
is indeed the case for [NiI(OEiBC)]−, by comparison of
Figure 1 to the published X-band spectrum (see Figure 12 in Renner et al.45), although careful EPR simulation
allowed these workers to extract the two components of g⊥: g = [2.061, 2.083, 2.2025], which values are
essentially identical to those obtained from 35 GHz spectra:
g = [2.063, 2.080, 2.204].50 In contrast, the 35 GHz
spectrum of NiIF
430 is as axial in appearance as its X-band
spectrum (see Figure 3 in Holliger et al.94).
It is interesting that, despite the potentially very lexible F430 macrocycle54,95 with its vast variety of sidechains,
including fused lactam (B) and cyclohexanone (C) rings, and the differences among the nitrogen donors (one is not conjugated with the other three), and the presence of two different Ni-N distances as determined by EXAFS,40 the
EPR spectrum of NiIF
430 is rigorously axial (with g|| = 2.244,
g⊥ = 2.063) to within ± 0.002 in g value (ca. 1 mT at
35 GHz, g = 2.0). We suggest that this may be evidence that
the orientation of the in-plane components of the g matrix
(gx, gy) may be exactly bisecting the N-Ni-N bond angles,
so that an average value results. Single crystal studies of CuII complexes have shown that an orientation of g
x, gy non-coincident with the Cu-N bond vector can occur.96,97
The 12,13-diepimer of NiIF
430 was also investigated, but its
35 GHz EPR spectrum in our hands was indistinguishable from native NiIF
430 (not shown), although a very slight
difference between the native and diepimeric forms has been reported.94 We have found that different preparations
and/or slight differences in buffer/glassing agent of NiIF 430
and of MCRred1 give variations in g values (e.g., ± 0.005
in g||) that is on the order of that reported for the diepimeric
versus native forms.
X-band EPR (and lower frequencies), however, can reveal hyperine splitting that is not resolved at higher ields/frequencies. The X-band EPR spectrum reported
for [NiI(OEiBC)]− shows resolved hyperine coupling at g⊥ from the four, essentially equivalent, pyrrole/pyrroline
nitrogens (A(14N)g⊥ = 0.98 mT, 28 MHz),47 which is not
seen at 35 GHz. The EPR feature at g⊥ for [NiI(OEiBC)]− is
qualitatively very similar to that seen for CuII tetrapyrroles,
such as [Cu(TPP)]18 or [Cu(OEP)] (A(14N)
g⊥ = 42 MHz).
98
Use of even lower microwave frequencies than X-band, such as S-band (ca. 1 GHz) or L-band (3 GHz), might
provide even better resolution of the 14N hyperfine
splitting, as has been shown for CuII complexes by Hyde
and Froncisz.4 In the case of NiIF
430, the reported X-band
spectrum reveals only a hint of resolved hyperine coupling, although “massaging” of the data (Fourier-iltered second derivative presentation) did reveal hyperine coupling (A(14N)iso = 1.0 mT, 29 MHz).94 The X-band spectrum
of NiIF
430M (the organic-soluble, pentamethyl ester of
F430) does show barely resolved 14N hyperine coupling
with A(14N)
g⊥ = 0.95 mT, 27 MHz.
33 The narrow range of 14N hyperine coupling for these complexes indicates a
commonality in bonding amongst them.
2.4. X-band and 35 GHz EPR Spectra of MCRred1
An extensive discussion of MCR, with its many forms, both EPR-active and EPR-silent,24-26,99-101 is outside the
scope of this study. We present here EPR spectra only of the form that is correlated with enzyme activity, MCRred1,22,102 which resembles by EPR spectroscopy most
closely NiIF
430 and [NiI(OEiBC)]−.103 The 35 GHz spectrum
Figure 1. Experimental (dashed trace of pair (colored in online version)) and simulated (solid black trace of pair) 35 GHz EPR spectra of NiIF
430
in aqueous solution and of [NiIOEiBC]– in 2-Methf. The spectra were
recorded at 2 K using the dispersion mode under passage conditions; a numerical irst derivative is shown. The abscissa is in g value to facilitate
comparison between spectra recorded at different frequencies (35.035 for NiIF
430; [NiIOEiBC]- for 35.422 GHz). The simulation parameters for
NiIF
430 are g|| = 2.244, g⊥ = 2.063, W|| = 140 MHz, W⊥ = 90 MHz (single crystal Gaussian linewidths, hwhm); for [NiIOEiBC]−: g
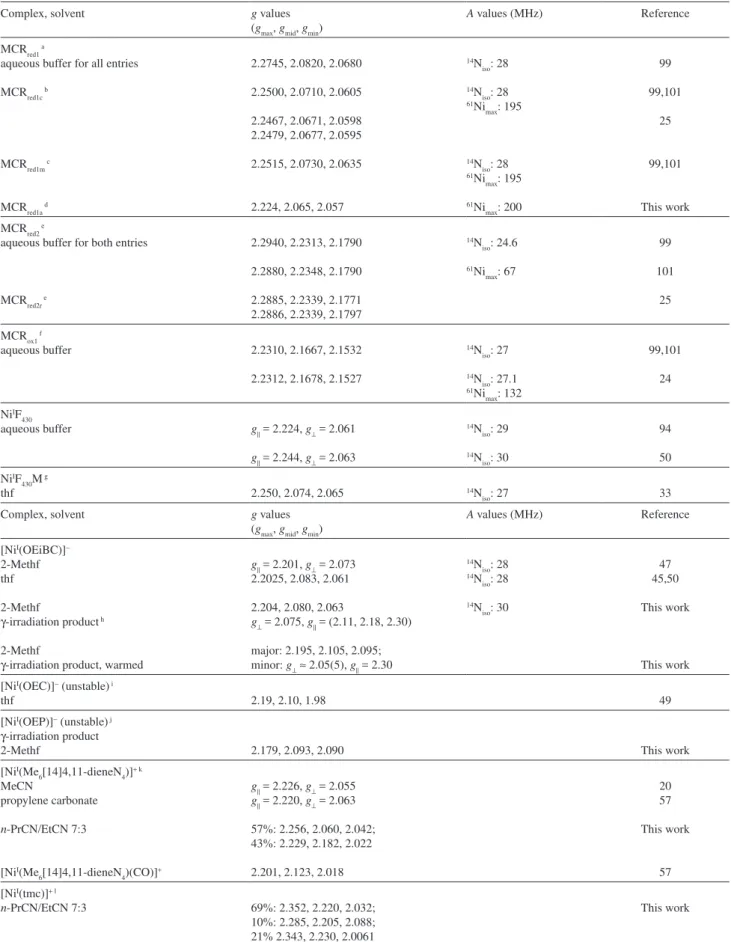
Table 1. Frozen solution EPR parameters for EPR-active MCR forms and tetraazamacrocyclic NiI complexes
Complex, solvent g values
(gmax, gmid, gmin)
A values (MHz) Reference
MCRred1 a
aqueous buffer for all entries
MCRred1c b
MCRred1m c
MCRred1a d
2.2745, 2.0820, 2.0680
2.2500, 2.0710, 2.0605
2.2467, 2.0671, 2.0598 2.2479, 2.0677, 2.0595
2.2515, 2.0730, 2.0635
2.224, 2.065, 2.057
14N iso: 28 14N
iso: 28 61Ni
max: 195
14N iso: 28 61Ni
max: 195
61Ni max: 200
99
99,101
25
99,101
This work MCRred2 e
aqueous buffer for both entries
MCRred2r e
2.2940, 2.2313, 2.1790
2.2880, 2.2348, 2.1790
2.2885, 2.2339, 2.1771 2.2886, 2.2339, 2.1797
14N iso: 24.6
61Ni max: 67
99
101
25
MCRox1 f
aqueous buffer 2.2310, 2.1667, 2.1532
2.2312, 2.1678, 2.1527
14N iso: 27
14N iso: 27.1 61Ni
max: 132
99,101
24
NiIF 430
aqueous buffer g|| = 2.224, g⊥ = 2.061
g|| = 2.244, g⊥ = 2.063
14N iso: 29
14N iso: 30
94
50 NiIF
430M g
thf 2.250, 2.074, 2.065 14N
iso: 27 33
Complex, solvent g values
(gmax, gmid, gmin)
A values (MHz) Reference
[NiI(OEiBC)]− 2-Methf thf
2-Methf
γ-irradiation product h
2-Methf
γ-irradiation product, warmed
g|| = 2.201, g⊥ = 2.073
2.2025, 2.083, 2.061
2.204, 2.080, 2.063
g⊥ = 2.075, g|| = (2.11, 2.18, 2.30)
major: 2.195, 2.105, 2.095; minor: g⊥≈ 2.05(5), g|| = 2.30
14N iso: 28 14N
iso: 28
14N iso: 30
47 45,50
This work
This work [NiI(OEC)]− (unstable) i
thf 2.19, 2.10, 1.98 49
[NiI(OEP)]− (unstable) j γ-irradiation product
2-Methf 2.179, 2.093, 2.090 This work
[NiI(Me
6[14]4,11-dieneN4)]
+ k
MeCN
propylene carbonate
n-PrCN/EtCN 7:3
[NiI(Me
6[14]4,11-dieneN4)(CO)]+
g|| = 2.226, g⊥ = 2.055 g|| = 2.220, g⊥ = 2.063
57%: 2.256, 2.060, 2.042; 43%: 2.229, 2.182, 2.022
2.201, 2.123, 2.018
20 57
This work
57 [NiI(tmc)]+ l
n-PrCN/EtCN 7:3 69%: 2.352, 2.220, 2.032;
10%: 2.285, 2.205, 2.088; 21% 2.343, 2.230, 2.0061
Complex, solvent g values (gmax, gmid, gmin)
A values (MHz) Reference
[NiI(Me
6[14]aneN4)]+
MeCN
propylene carbonate
[NiI(Me
6[14]aneN4)(CO)]+
propylene carbonate
g|| = 2.266, g⊥ = 2.055
g|| = 2.253, g⊥ = 2.054
2.198, 2.123, 2.012
20 57
57
Complex, solvent g values
(gmax, gmid, gmin)
A values (MHz) Reference
[NiI(tmc)(O 2)]
+
dmf/toluene 1:2 2.29, 2.21, 2.09 108
a Red1 EPR signal present in the MCR
red2 sample showing a mixture of red2 and red1 signals;99 in the red2 form, the NiI is coordinated by the thiol(ate)
sulfur of coenzyme M (HSCoM = HSCH2CH2SO3−).36b Red1 EPR signal in the presence of coenzyme M; this signal is referred to as MCR
red1c. c Red1
EPR signal in the presence of methyl-coenzyme M (CH3SCH2CH2SO3−); this signal is referred to as MCR
red1m. d Red1 EPR signal in the absence of other
substrates, coenzymes, or other forms of MCR. This signal is denoted MCRred1a, but simulation parameters for this speciic form could not be found in the relevant references of Thauer and co-workers.99-101e Red2 signal as originally reported; this is now referred to as MCR
red2r (r = rhombic); there is also an
axial red2 signal denoted MCRred2a, with EPR signals very similar to MCRred1a.25 Slightly different EPR parameters result depending on the two methods
of generation of MCRred2r;25 both parameter sets are given here. f Ox1 signal as originally reported. Harmer et al. determined the full A(14N) tensor for all
four nitrogens of the macrocycle;24 the average, isotropic value of them all is given here for comparison with less reined data. In MCR
ox1, there is formally
a NiIII ion with thiolate ligation from CoM [NiIII-(RS−)]; this can also be considered as a spin-coupled NiII -thiyl system, [NiII-(RS•)].64g Pentamethyl ester
of F430.33h (2,3,7,8-tetrahydro-2,3,7,8,12,13,17,18-octaethylporphyrin) NiII (octaethylisobacteriochlorin) cryoreduced by γ-irradiation; the resulting NiI
species is heterogeneous, with several features that can be assigned to g||. The second entry is for the sample after brief (ca. 5 s) warming to 300 K under nitrogen. i (2,3-dihydro-2,3,7,8,12,13,17,18-octaethylporphyrin) NiII (octaethylchlorin) chemically reduced by sodium tetracenide; the resulting NiI species
is unstable towards formation of a chlorin-phlorin anion.49j (2,3,7,8,12,13,17,18-octaethylporphyrin) NiII cryoreduced by γ-irradiation; the NiI signal
disappears upon brief warming to 300 K under nitrogen. k Mixture of meso and rac forms of
(5,7,7,12,14,14-hexamethyl-1,4,8,11-tetraazacyclotetradeca-4,11-diene) NiI perchlorate.58 The relative amount of the two isomers is unknown. l Mixture of RRSS and RSRS isomeric forms of
(1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) NiI (tmc) triluoromethylsulfonate. The relative amount of the two isomers is unknown, but is ca. 3:1 at equilibrium
in aqueous solution.78m (C-meso-1,4,5,7,7,8,11,12,14,14-decamethyl-1,4,8,11-tetraazacyclotetradecane) NiII (Me
6[14]ane(NMe)4, Me6tmc) reduced by
steady-state radiolysis.79
Table 1. Frozen solution EPR parameters for EPR-active MCR forms and tetraazamacrocyclic NiI complexes (cont.)
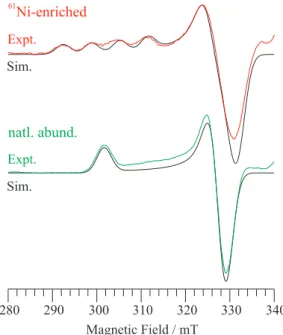
of MCRred1 is shown in Figure 2, for both natural isotopic abundance and 61Ni-enriched samples. The EPR spectrum
of the natural-abundance sample is almost the same as that for NiIF
430, with g = [2.224, 2.065, 2.057], indicating
that the electronic structure about the NiI ion, including
the nearly axial symmetry, is the same in the protein as in the isolated cofactor. In the enriched enzyme sample, hyperine splitting due to 61Ni (I = 3/2) is apparent at g
||,
but is essentially unobservable at g⊥. The use of 61Ni thus
yields an EPR spectrum for MCRred1 that resembles that for typical tetragonally distorted six-coordinate (square pyramidal, sq pyr) CuII (63,65Cu, I = 3/2, 100% abundance)
with dxz,yz4dxy2dz22dx2–y21coniguration. The natural abundance
35 GHz spectrum reveals very slight rhombic symmetry at g⊥, but the resolution of the hyperine splitting at g|| in
the enriched sample is less than ideal. However, X-band EPR provides good resolution of A(61Ni)
|| = 200(10) MHz,
equivalent to the value reported elsewhere.99,101 This
result is analogous to the improved resolution at X-band compared to 35 GHz of 14N hyperine described above
for NiIF
430 and [NiI(OEiBC)]−. The chief difference
between the 61Ni-enriched spectrum for MCR red1 and
that for typical CuII tetrapyrroles is the larger magnitude
hyperine coupling in the latter (e.g., A(63Cu)|| = 630 MHz
for [Cu(OEP)]98). This three-fold larger A value in the CuII
complex is largely the consequence of the three-fold larger
gN for Cu (63Cu, g
N = 1.484; 65Cu, gN = 1.588) versus61Ni
(gN = −0.500),5 so that the bonding in the two complexes
is actually quite similar (A/gN = 400 for 61Ni; 424 for 63Cu).
Indeed, the M-N bond distances are also quite similar. The CuII-N distances in [Cu(OEP)] are 199.6(3) pm
(Cu-N(1)) and 199.9(3) pm (Cu-N(2)),104 while the
EXAFS-determined NiI–N distances in [NiI(OEiBC)]− are
two at 191 pm and two at 207 pm,45 and for NiIF 430M,
two at 188 pm and two at 203 pm.40 It is unfortunate that
cost precludes 61Ni hyperine coupling data from being
more available for NiI complexes in general, but the EPR
results for MCRred1 clearly show that this species, and by extension, isolated NiIF
430, whether in aqueous or organic
solvent, and [NiI(OEiBC)]−,are all typical tetragonally
distorted (whether square planar (sq pl), square pyramidal, or even six-coordinate, is not signiicant) dxy,xz,yz,z28dx2–y21
complexes, such as commonly found for CuII with
2.5. 35 GHz EPR spectra of [NiI(Me
6[14]4,11-dieneN4)]
+
The complex [NiI(Me
6[14]4,11-dieneN4)](ClO4)
represents a step away from π-conjugation relative to the species discussed above. It is one of the few NiI
complexes to be crystallographically characterized (in the racemic form) and was thus used for bond distance calibration in EXAFS studies of NiI species for which no
crystal structures were available (e.g., NiIF430).58 It is also
representative of the many tetraaza macrocyclic complexes described by Busch and co-workers.20 The X-band spectrum
of this electrochemically generated complex in propylene carbonate frozen solution was reported by Gagné and Ingle57 and gave g
|| = 2.220, g⊥ = 2.063. As can be seen
from Table 1, these values are totally unremarkable, and indeed, are almost the same as those for the NiI species
described in the preceding sections. Here, however, a mixed nitrile solvent system was used (n-butyronitrile/
propionitrile, 7:3 v/v), which provides a good glass for EPR. This nitrile solvent system is effective at dissolving the ionic complex and nitriles would be expected to be relatively weak donors, compared to, e.g., CO, the binding
of which had been extensively studied in NiI complexes.57
Nevertheless, an EPR spectrum quite different from that of the NiI species discussed hitherto results, as shown in
Figure 4. The signal is clearly heterogeneous, and can be adequately described as the superposition in roughly equal amounts of two signals, one described by g = [2.256,
2.060, 2.042], and one with g = [2.229, 2.182, 2.022]. The
former, nearly axial g matrix, while different from those
previously reported,20,57 is nevertheless similar to that for the
other tetragonal NiI complexes described herein (Table 1).
The better ield dispersion of 35 GHz EPR might allow resolution of rhombic splitting that was not observable in the earlier X-band studies,20,57 and the difference in solvent
might be responsible for the other differences – note the variation in g values among the various forms of MCRred1
and of NiIF
430 – all in aqueous solvent (Table 1). Note
also that the crystal structure of meso-[NiI(Me6
[14]4,11-dieneN4)](ClO4) shows a planar NiN4 unit with two sets of Ni-N bond distances,58 which would be expected to give a
slightly rhombic, tetragonal type (g||ca. 2.2 > g⊥ca. 2.05)
of EPR signal. The EPR signal with the axial g is thus
assigned to a typical, tetragonal NiI tetraaza macrocycle:
dxz,yz4dxy2dz22dx2–y21 ; sq pl in the absence of any axial ligand
(from solvent) coordination; ive-coordinate with one axial ligand; six-coordinate with two, all analogous to CuII
complexes of the same geometry.
What about the rhombic signal? Such a signal is similar to that seen for MCRred2: g = [2.2940, 2.2385, 2.1790].99
In this MCR form, there is an axial sulfur donor (from Figure 2. Experimental (dashed trace of pair (colored in online version))
and simulated (solid black trace of pair) 35 GHz EPR spectra of MCRred1 in natural isotopic abundance and 61Ni-enriched. The spectra were recorded
at 2 K using the dispersion mode under passage conditions; a numerical irst derivative is shown. The abscissa is in g value to facilitate comparison
between spectra recorded at different frequencies (35.035 for natural abundance; 34.945 GHz for 61Ni-enriched). The simulation parameters
are g = [2.224, 2.065, 2.057], W = 90 MHz (single crystal Gaussian linewidths, hwhm); the enriched sample includes: A(61Ni)g
max = 200 MHz.
Figure 3. Experimental (dashed trace of pair (colored in online version)) and simulated (solid black trace of pair) X-band (9.47 GHz) EPR spectra recorded at 77 K of MCRred1 in natural isotopic abundance and
coenzyme M) to the NiI ion.25,36 Perhaps more relevant,
exposure to CO leads to formation of [NiI(Me
6
[14]4,11-dieneN4)(CO)]+ with g = [2.201, 2.123, 2.018].57 One
could propose therefore, that the highly rhombic signal observed for [NiI(Me
6[14]4,11-dieneN4)]+ results from
axial coordination by a nitrile involving π-donation from NiI to the axial ligand, as with NiI−CO bonding. That a
nitrile could have this effect would be a statement as to the powerful π-donor abilities of NiI, which is related to
its nucleophilic role in MCR action. However, previous EPR studies on NiI complexes showed no such behavior
in acetonitrile solvent.20 It is apparently the case here
that the lexible macrocyclic ligand, whether the cause or effect of nitrile binding, adopts a conformation that is highly distorted from square planar tetraaza (overall square pyramidal due to one axial nitrile, CO, thiol(ate) etc), becoming trigonal bipyramidal (tbp) in the extreme case. For ideal tbp geometry, which for d9 has the electronic
conigurationdxz,yz4dxy,x2–y24dz21, the g values are: g
||≅ 2.00 <
g⊥≅ 2.25(5).105,106 Such an axial signal is not seen here,
but the lower symmetry present in these NiI complexes is
unlikely ever to yield ideal tbp geometry.
What about intermediate geometries? This situation is much more complicated, but has been beautifully worked out using ligand-ield theory by Bencini and co-workers.105,106 This theoretical work was in conjunction
with their EPR studies on bis(N-methylsalicylaldiminato)
complexes of CuII, which quinquidentate ligand strongly
favors tbp coordination geometry. Bencini et al.105,106
provided equations for the g tensor components for the
entire transition from square pyramidal to tbp in C2v
symmetry. They explain (especially see Figure 4 in Bencini
et al.106) that this change causes gmax (gz) slightly to decrease
from roughly 2.30 to 2.20; gmin (gx) likewise decreases also
only slightly, from 2.07 to 2.00; however gmid (gy) varies
signiicantly during this transition, from roughly 2.07 to 2.22.
Such a geometry that approaches tbp could thus be proposed for the second species in [NiI(Me
6
[14]4,11-dieneN4)]+, that with g = [2.229, 2.182, 2.022]; this g
could correspond approximately to αca. 115o, where 90o
(sq pyr) ≤α≤ 120o (tbp). Equations for A(63,65Cu) were also
given,106 which should be applicable to 61Ni. Unfortunately,
there are no A(61Ni) data to which to apply the Bencini
model except for MCRred1, which its their model of a typical tetragonal/square pyramidal system. One would expect that
61Ni-enriched [NiI(Me
6[14]4,11-dieneN4)]+ would show
large (ca. 200 MHz) 61Ni hyperine coupling at g
max (gz) for the axial (sq pyr) signal and smaller for the rhombic (tbp) signal. We further speculate that the rac form corresponds
to the rhombic EPR signal, as this form binds CO,57,58 while
the meso form corresponds to the axial EPR signal, similar
to the structurally characterized form.
2.6. 35 GHz EPR spectra of [NiI(tmc)]+
The final, stable NiI species to be described here
is that with the fully saturated tetraaza macrocyclic ligand, tmc. The crystal structure of RRSS-[NiI(tmc)]
(CF3SO3)•NaCF
3SO3 shows that the geometry around
the NiI ion is exactly planar with two sets of Ni−N bond
distances (209.5 and 212.0 pm),76 analogous to the results
for [NiI(OEiBC)]−. The EPR spectrum of [NiI(tmc)]+ has
not, to our knowledge, been previously reported. However, the spectra for other fully saturated tetraaza macrocyclic complexes of electrochemically generated NiI have
been reported, such as with Me2[14]aneN4 (g|| = 2.261, g⊥ = 2.060) and Me6[14]aneN4 (g|| = 2.266, g⊥ = 2.055 in
acetonitrile solution; g|| = 2.253, g⊥ = 2.054 in propylene
carbonate solution).20,57 These EPR parameters are again
very similar to many other such tetragonal/sq pyr/sq pl complexes (see Table 1).
In contrast to these clear-cut, earlier results, the 35 GHz EPR spectrum of [NiI(tmc)]+ in the nitrile solvent system is
heterogeneous. As shown in Figure 5, the spectrum can be deconvoluted into at least two components, or better with three. Two components are expected since, although the solution was prepared from solid RRSS isomer, in solution there is
Figure 4. Experimental (dashed trace (colored in online version)) and simulated (solid black trace) 35 GHz EPR spectrum of [NiI(Me
6
[14]4,11-dieneN4)](ClO4) in n-butyronitrile/propionitrile (7:3 v/v) frozen solution. The spectrum was recorded at 2 K (and 35.116 GHz) using the dispersion mode under passage conditions; both the experimental absorption lineshape (lower dashed (colored) trace) and a digital irst derivative (upper dashed (colored) trace) are shown with their simulations. The simulation is the sum of two parameter sets: 57% integrated intensity weighting using g = [2.256, 2.060, 2.042], W = [100, 60, 60] MHz (single crystal Gaussian linewidths,
inter conversion so that the RSRS isomer is also present.76,78
The relative amount of the two isomers in nitrile solutions is unknown, but at equilibrium is ca. 3:1 in aqueous solution,78
so that the deconvolution into 69% major component and 31% two minor components is not that far off from the aqueous solution result. However, these three EPR components are all highly rhombic and none resembles typical tetraaza macrocyclic complexes of NiI (i.e., CuII-like parameters:
g||≅ 2.25(5), g⊥≅ 2.05(5); see Table 1), as would be expected from the crystal structure. A possible explanation is that axial coordination of the nitrile ligand leads to formation of species that are electronically very similar to the CO adducts of the NiI
macrocycles reported by Gagné and Ingle.57 This possibility
was raised above to explain the rhombic component in the EPR spectrum of [NiI(Me
6[14]aneN4)]+ (Figure 4). It is surprising,
however, that butyronitrile/propionitrile would behave as the strong π-acceptor CO does. Furthermore, although Gagné and Ingle used the polar, but totally non-coordinating solvent propylene carbonate,57 acetonitrile was employed earlier by
Lovecchio, Gore, and Busch,20 and their spectra differ only
trivially from the corresponding spectra reported by Gagné and Ingle.57 The rhombic signals seen for [NiI(tmc)]+ also
strongly resemble those for MCRred2,36,99 however, these result
from an axial thiolate ligand (from coenzyme M) to NiI, and
no such species is available in the present case. Meyerstein and co-workers79,80 used radiolysis, as well as electrochemistry,
to generate tetraaza macrocyclic complexes of NiI from NiII
in aqueous solution. They reported EPR spectra at 77 K of radiolytically generated [NiI(Me
6tmc)]+ that were typical for
a tetragonal complex (g|| = 2.333, g⊥ = 2.069; see Table 1);
however, in the presence of formate ion, highly rhombic spectra were observed: g = [2.261, 2.136, 2.073].79 No
explanation for this was given.
Our speculation for the EPR behavior of [NiI(tmc)]+ in
nitrile frozen solution, and possibly the results of Jubran et al.,79 is the same as that given above for [NiI(Me6
[14]4,11-dieneN4)]+, namely that there is distortion away from sq
pl or sq pyr (with axial nitrile) geometry towards either distorted tetrahedral or tbp geometry (with equatorial nitrile), which leads to mixing in of dz21 character into the
ground state. The difference among the three forms seen by EPR is relatively slight; we can only speculate the one form corresponds to one isomer in a given geometry, whether distorted tetrahedral or tbp (due to nitrile coordination), and the other two to the other isomer in each of these geometries (or tbp with both axial and equatorial nitrile coordination).
2.7. Discussion of “nickel(I)-dioxygen” species
Solution samples of the NiI complexes that were
provided as solids, meso, rac-[NiI(Me
6[14]aneN4)]+ and
RRSS-[NiI(tmc)]+, were prepared under inert atmosphere.
However, the possibility that some amount of dioxygen adducts were formed cannot be totally excluded. We therefore summarize here very interesting and recent studies by Riordan and co-workers107 on dioxygen binding
to NiI complexes, including RRSS-[NiI(tmc)]+ in a variety of
solvents (e.g., MeCN, thf, dmf and MeOH).108 A complex
they formulated as [Ni(tmc)(O2)]+ exhibited a rhombic EPR
signal (X-band, 14 K) with g = [2.29, 2.21, 2.09],108 which
is remarkably similar to those for CO adducts of NiI tetraaza
macrocycles.57 However, a wide variety of other physical
techniques were used to characterize this dioxygen complex in solution, including UV-Vis, XAS, and Resonance Raman spectroscopy. Such a species can have multiple descriptions: [NiI-O
20]+ (dioxygen), [NiII-O2−]+ (superoxo),
or [NiIII-O
22−]+ (peroxo), which we will evaluate here.
The NiIII-peroxo formulation would be expected to
give EPR spectra typical of such low-spin 3d7 complexes
(for tetraaza macrocyclic complexes of NiIII: g
||≅ 2.02(2),
g⊥ ≅ 2.20(2)20,109), which is similar to that of [Ni(tmc)
(O2)]+ (g
|| = 2.09, g⊥ = 2.25(4)). The EPR spectrum of
[Ni(tmc)(O2)]+ is optimal at low temperature (6 K) and
decreases with higher temperature (see Figure S9 in Kieber-Emmons et al.108). In contrast, EPR spectra for
authentic NiIII complexes are readily observed even at
room temperature.109 This suggests to us that the NiIII
-peroxo description (which was disfavored based on other Figure 5. Experimental (dashed trace (colored in online version)) and
simulated (solid black trace) 35 GHz EPR spectrum of RRSS-[NiI(tmc)]
(CF3SO3)•NaCF
3SO3 in n-butyronitrile/propionitrile (7:3 v/v) frozen
solution. The spectrum was recorded at 2 K (and 35.198 GHz) using the dispersion mode under passage conditions; both the experimental absorption lineshape (lower dashed (colored) trace) and a digital irst derivative (upper dashed (colored) trace) are shown with their simulations. In the left panel, the simulation is the sum of two parameter sets: 87% integrated intensity weighting using g = [2.352, 2.220, 2.032], 13% weighting using g = [2.285, 2.205, 2.088], for both W = [100, 80, 60] MHz (single crystal
 2 ; CSD code:](https://thumb-eu.123doks.com/thumbv2/123dok_br/18994062.461631/7.892.125.401.108.269/diagram-shown-showed-stability-structure-form-complex-reported.webp)
![Figure 1. Experimental (dashed trace of pair (colored in online version)) and simulated (solid black trace of pair) 35 GHz EPR spectra of Ni I F 430 in aqueous solution and of [Ni I OEiBC] – in 2-Methf](https://thumb-eu.123doks.com/thumbv2/123dok_br/18994062.461631/8.892.495.759.113.368/figure-experimental-colored-version-simulated-spectra-aqueous-solution.webp)

![Figure 4. Experimental (dashed trace (colored in online version)) and simulated (solid black trace) 35 GHz EPR spectrum of [Ni I (Me 6 [14]4,11-dieneN 4 )](ClO 4 ) in n-butyronitrile/propionitrile (7:3 v/v) frozen solution](https://thumb-eu.123doks.com/thumbv2/123dok_br/18994062.461631/12.892.522.730.103.411/figure-experimental-colored-simulated-spectrum-butyronitrile-propionitrile-solution.webp)
![Figure 5. Experimental (dashed trace (colored in online version)) and simulated (solid black trace) 35 GHz EPR spectrum of RRSS-[Ni I (tmc)]](https://thumb-eu.123doks.com/thumbv2/123dok_br/18994062.461631/13.892.97.437.114.355/figure-experimental-dashed-colored-online-version-simulated-spectrum.webp)
![Figure 7. Experimental (dashed trace (colored in online version)) and simulated (solid black trace) EPR spectrum of ttt-[Ni II (OEiBC)] in 2-Methf solution γ-irradiated at 77 K and then warmed to 300 K for 5 s under N 2 atmosphere](https://thumb-eu.123doks.com/thumbv2/123dok_br/18994062.461631/15.892.134.396.114.445/figure-experimental-colored-simulated-spectrum-solution-irradiated-atmosphere.webp)
![Figure 8. Experimental (dashed trace (colored in online version)) and simulated (solid black trace) EPR spectrum of [Ni II (OEP)] in 2-Methf solution γ-irradiated at 77 K](https://thumb-eu.123doks.com/thumbv2/123dok_br/18994062.461631/16.892.111.364.108.519/figure-experimental-colored-version-simulated-spectrum-solution-irradiated.webp)
