RISK AS A REGULATORY TOOL
Abstract
The development of regulations and its application by regulators is moving from a prescriptive approach to a more risk-based one. This work looked at how risk has been used in regulation and how effective has it been its application, in other words how valid is the use of risk as a regulatory tool. A search was made on if and how regulators of different areas have been using risk management. Afterwards a set of public informa-tion from a regulator was analysed to find how such an analysis could contribute to a risk-based approach to regulatory action. Findings have shown that although in different stages, tendency in different fields is to increase the use ok risk as a regulatory tool to improve regulatory effectiveness.
Keywords: Risk, risk-based regulation, regulatory tool, risk assessment, risk management, resources.
Resumo
A elaboração de novos normativos legais e a sua aplicação por reguladores tem vindo a afastar-se de uma abordagem prescritiva no sentido de uma abordagem baseada no risco.
Este trabalho analisa a forma como o risco tem vindo a ser utilizado na regulação e quão efetiva tem sido a sua aplicação, ou seja, que validade tem a utilização do risco como uma ferramenta regulamentar. Foi efe-tuada uma pesquisa bibliográfica sobre se, e o modo como, a gestão do risco tem vindo a ser utilizada por reguladores de diferentes áreas. Procedeu-se em seguida a uma análise de informação publicamente dispo-nibilizada por um regulador, com o objetivo de determinar como tal análise poderia contribuir para uma abordagem da atividade regulamentar baseada no risco. Apesar de se encontrar em diferentes níveis de uti-lização, a tendência em diferentes áreas tem sido a de aumentar a utilização do risco como uma ferramenta regulamentar para melhorar a efetividade da ação reguladora.
Palavras-chave: Risco, regulação baseada no risco, ferramenta regulamentar, avaliação do risco, gestão do
risco, recursos.
Rui Vilar
Mestre integrado em Ciências Farmacêuticas, Faculdade de Farmácia da UL.
Rui Loureiro
Professor da Faculdade de Farmácia da Universidade de Lisboa.
Introduction
Regulators face increasing demands for improved
outcomes under limited resources. Risk-based
meth-odologies are known to deal with resource manage-ment to improve results under limited resources. This work looks at finding if regulators are using risk methodologies and if risk could be considered a valid regulatory tool.
Methodology
To encounter uses of risk in regulation, a literature search was conducted using the search terms regula-tion, risk, risk-based, hazard, deterministic, decision, supervision, inspection, GMP, industry, control, per-formance, sampling, management, nuclear, oil and
gas and pharmaceutical. Search was made in Google and in Google Scholar, PubMed, b-on databases be-tween March and May 2010.
Use of Risk in Regulation
Governments and regulators have encouraged the use of risk in the review, development and imple-mentation of new, better regulation to answer more efficiently to regulatory concerns.
Risk-based regulation is regarded as less inefficient than traditional regulation, because of the allocation of resources in proportion to risks1-3. Through the use of risk-assessment, regulator bodies can understand the nature of businesses, processes, products and
outcomes, and external factors affecting their risks to
focus available resources on highest risk businesses, 36
processes, products and outcomes, and least on those
with best records of compliance. Overall the inten-sity of a regulator body oversight should be adjusted to the degree of assessed risk.
The Australian government, the European Union and the United Kingdom have encouraged use of risk reg-ulation to improve competitiveness4-7. Two-thirds of the Organization for Economic Cooperation and De-velopment (OECD) countries reported some require-ment for risk assessrequire-ment8.
The main feature of risk-based regulation is the al-location of resources according to risk of products, processes and outcomes, to increase effectiveness of
regulation.
Use of this main feature can be found in the use of tier sys-tems for licensing, monitoring, and reviewing, allocating resources in inspections and differentiating fee charging according to risk, by environmental agencies9-19. Medicines regulatory bodies have also implemented risk-based inspections20-29. Effective use of inspec-tion resources is seen as benefiting not only regulator bodies but also manufacturers30. Additionally, as an answer to the overwhelming of medicines regulatory bodies´ resources, due to increasing new applications and supplements, these regulators have adopted new risk-based regulations and risk-based review sys-tems:
• An annual reporting system of minor variations that requires no prior approval allows regulators to focus on variations with greater impact on quality, safety and efficacy (the underlying regulation follows in itself a risk-based classification of variations)31-32;
• Change management protocols allow for
implemen-tation of major variations as if a minor variation31-32;
• Risk-based comparability protocols allow for the
implementation of variations without prior review or approval by FDA33-34.
• Use of design spaces (working within a design space would not be considered a change)31-32.
• FDA has developed question based review (QbR) for abbreviated new drug applications (ANDAs), a risk-based pharmaceutical quality assessment sup-ported in quality by design (QbD), aimed at focus-ing assessment resources on critical pharmaceuti-cal quality attributes, and shifting them from low risk products35-37. It substituted quality by testing review (QbT) assessment where all products were treated equally without regard of the risk to final consumers.
Use of QbD and design spaces is associated with an-other feature of risk use, which is the need for tech-nical and scientific developments for the adoption
of risk-based approaches. Risk-based reduction of oversight by regulators is possible due to the level of product and process understanding that is achieved with the use of QbD and design spaces. Recent works considered use of risk methodologies central in devel-opment of QbD and design space. In manufacturing the end objective of risk methodologies would be to keep production at a low risk state36,38-41.
A risk-based culture in medicines regulation is encour-aged by ICH Q8, Q9 and Q10 guidelines42-44. Such incen-tive is made for development, manufacturing and dis-tribution43. We consider that this should also include safe disposal of medicines, and in a holistic approach services and information. Concerns expressed by these
guidelines are the use of risk methodologies to prevent
blocking of continuous manufacturing improvements, adoption of new technologies and allow real time as-surance of quality.
Technical and scientific developments were fundamen-tal for the adoption of risk regulation in the nuclear in-dustry. In this case knowledge of structures, safety sys-tems and its degradation processes, necessary for risk analysis, allowed the adoption of risk-informed regu-lations consisting of a mixture of both risk-based and deterministic regulations45-46. Interest in risk-based regulation arose when probabilistic risk assessment (PRA) studies revealed safety concerns despite confor-mance with the deterministic “defence-in-depth” con-cept. Risk-informed approaches are also considered for upcoming advanced nuclear reactors1,46-48. As regards management of resources in the nuclear industry, risk analysis could improve the risk and cost concerns of nuclear power plants49.
As noticed with nuclear industry, another feature asso-ciated with risk-based regulation is increase in safety. Safety concerns are the drive for use of risk in vigilance activities, an important regulatory activity, as is the case with risk-based veterinary surveillance (e.g. tar-geted surveillance of bovine spongiform encephalopa-thy), and pharmacovigilance of drug products50. The risk-based strategy developed in the EU for pharma-covigilance mainly concentrated in the introduction of risk management plans (EU-RMP)51-54. These are mandatory in some marketing authorization applica-tions and entail the basic risk management cycle55-57. In pharmacovigilance risk management has the ob-jective of keeping favourable the risk-benefit balance. Recently the new pharmacovigilance legislation58-59 introduced new features that strengthen the use of risk in pharmacovigilance. Risk-management plans are now applicable to all types of marketing authorization, though proportionate to identified and potential risks of the medicinal product, and the need for
risation safety data. The broadening of the adverse re-action definition enlarges the scope of risks associated with medicines in pharmacovigilance. The differences in frequency and exemptions in the European Union reference dates and frequency of submission of period-ic safety update reports (PSUR), reveals the existence of different levels of risks and better resource alloca-tion concerns. Single assessment enforces concentra-tion of available data to risk-benefit analysis initiated by PSUR worksharing. Gathering of information is an important aspect of risk methodology.
Regulating and implementing regulations has risks. These risks should be considered in use of risk-based regulation. Use of risk-based approaches to develop better regulations is encouraged to reduce incidence of failure in regulating, which may come from regulating when there is no need or failing to regulate when there is need60-61. In implementing regulations, risk-based regulation acknowledges that compliance with targets set in regulations cannot be fully enforced, a gap that has been called “risk gap”. Targets may be unobtain-able, not necessary or not justifiable. This is the case with zero risk policies: assessment of risks involved may conclude that reducing risk to zero requires unjus-tifiable resources and that a certain level of risk can be acceptable considering benefits4,62. When using risk- -based regulation regulatory bodies should be aware of the risk of failing to improve regulatory outcomes, if risk
regulation is seen as the assessment and management of institutional risks instead of the risks to society63.
A Risk-Analysis
As seen, risk-based regulation has been introduced in different regulatory areas. A practical approach could contribute to better understand the use of risk-based regulatory action.
The main elements of a risk-based framework for regulation are setting an organization risk-tolerance, risk identification and risk assessment, assigning scores and ranking firms or sites (in our view there
should also be ranking of products, processes, and a
mixture of all)64. To do so information held by regu-latory bodies has to be analysed using a risk perspec-tive. We have used the Portuguese National Author-ity of Medicines and Health Products (INFARMED, I.P.) out of stock medicines list, quality alerts and safety alerts, published in its webpage, to see how this is within reach of regulatory bodies and if such information could be used as a regulatory tool in de-cision-making65-68.
The out of stock medicines list consists of a software application that should be filled in by marketing au-thorization holders (MAH) whenever it is expected
that a medicine will be out of stock for at least fif-teen days. The list contains only medicines subject to medical prescription.
The objectives behind the development of this list were to inform health professionals and citizens on out of stock medicines.
A codification system was applied to reasons provid-ed by MAH for the out of stock of mprovid-edicines in the list, as identified in Table I. Different strengths of the same medicine were considered only one medicine to take account for the existence of medicines with sev-eral strengths.
Table I – Codification of reasons provided by Marketing Authorization Holders (MAH) for the out of stock of medicines (data from April 2005 to 12th of June 2010)
Ceased marketing A 83 22,7
Code Frequency
(n=366)
Percentage Reason
No reason stated and no prevision for reposition B 231 63,1 Marketing authorization ceased to be valid C 5 1,4 New manufacturer due to small dimension of market D 2 0,5 New formulation E 2 0,5 No reason stated, with prevision date for reposition F 31 8,5 MAH transfer G 2 0,5 Name change H 1 0,3 Seasonal vaccine I 2 0,5 Delay in manufacturing J 2 0,5 Decision on reimbursement K 1 0,3 Awaits approval of variations by regulator L 2 0,5 Awaits price review M 1 0,3 No reason stated; already on market N 1 0,3
A Pareto analysis (Graphic 1) showed that classes A and B account for the majority of out of stock medi-cines (85,8%). In most of the cases (71,6%) MAH didn’t provide any reason for the out of stock of the medi-cines (classes F and B). This precludes an analysis of what actions the regulator body could take, however a deeper investigation of the causes is warranting if the regulator body chooses to evaluate if and which mea-sures could be adopted. This should be the case as it concerns impacts on health. Experience gained in the investigation of the causes could allow for a standard selection list of reasons (without the no reason option),
associated with a codification system as exemplified in this work.
70 60 50 40 30 20 10 0 63,1 22,7 8,5 1,4 0,5 0,5 0,5 0,5 0,5 0,5 0,3 0,3 0,3 0,3 Percentage Justifications Reasons for the out of stock of medicines provided by Marketing Authorization Holders Graphic 1 – Pareto analysis of the classes of reasons for out of stock of medicines, as identified in Table I
Note: Mostly MAH do not provide any justification or date for re-supply. Adding A, B and F the total percentage is 94,3%.
Another useful information would be to mention the due date for market re-supply. This date was men-tioned in only 8,5% of the cases. The time during which the medicine is out of stock could be related to the risk of its unavailability and thus to actions by the regulator body. Thus, a better specification of the reasons and a prediction of the re-supply date, as time would be related to impact of the medicine being out of stock, would allow for a better risk analysis and decision-making on actions the regulator body could adopt. A simple example can be withdrawn from the data set. There were two medicines out of stock due to variations pending approval. In one case there was no therapeutic alternative. The identification of this risk in a risk analysis should alert the regulator body to end this situation by allocating resources to the as-sessment of the variations.
Analysis of the five-year data of the list (Graphic 2) showed an increase in the number of out of stock medicines along the years since 2005. This tendency could be expected also for 2010, as numbers in first semester were already similar to total for 2009. To this observation could be contributing the newly implementation of the system, thus a general conclu-sion that market problems have been rising cannot be taken. This could be partially what was already happening in the market but was not being moni-tored. However, the system has indeed a history of five years and a doubling on the number of out of stock medicines in 2010, as compared to 2009, if the same tendency is registered in the second semester of 2010, is a sharp rise that could not be explained by a sudden adherence of MAH to the system. If the same tendency were to be observed until end of 2010, a sharp rise on the out of stock of medicines should
be deemed a careful study. The annual increase on the number of out of stock medicines is observed for gener-ic and non-genergener-ic medgener-icines (Graphgener-ic 3). Also, as seen in Graphic 2, numbers in mid 2010 were again similar to total for 2009 in generic and non-generic groups. The slope of the curve for generic medicines was greater between 2007 and 2009, year in which it ex-ceeded the number of non-generic medicines. This is possibly linked to the development of a generic medicines market in Portugal69. Accounting only for these figures, if initially risk of the existence of out of stock medicines was greater with non-generic medi-cines, since 2009 risk is similar for both generic and non-generic medicines. This means that from a risk perspective the same level of resources should be al-located for both groups. However, when accounting also for the existence of a therapeutic alternative, risk seems different for both groups. Data on existence of a therapeutic alternative is available from the out of stock medicines list. Graphic 4 plots this for generic and non-generic medicines. In 92% of the cases there was a therapeutic alternative when the out of stock medicine was a generic. With non-generic medicines only in 50% of the cases a therapeutic alternative was available. Thus, if initially risk seemed similar for both groups, impact of out of stock non-generic medicines is higher than for generic medicines. Then from a risk perspective the level of resources allocat-ed to deal with out of stock non-generic mallocat-edicines should be higher than for generic medicines.
77 25 55 75 39 10 18 5 3 6 33 20 102 108 75 45 21 15 0 20 40 60 80 100 120 2005 2006 2007 2008 2009 2010 (January -12th June) Year Number of medicines
Sterile Non-sterile Total Graphic 2 – Annual variation in the number of out of stock medicines (total, sterile and non-sterile medicines)
Regarding quality alerts a classification system based on the reason that deployed the quality alert was de-veloped as presented in Table II. The main reasons for quality alerts were problems with specifica-tions, primary packaging and stability of medicines.
16 48 29 54 14 35 46 53 1 5 10 55 0 10 20 30 40 50 60 2005 2006 2007 2008 2009 2010 (January -12th June) Year Number of medicines Non-generic Generic
Graphic 3 – Annual variation in the number of unavailable medicines (generic and non-generic medicines)
50 8 50 92 0 10 20 30 40 50 60 70 80 90 100
Non-generic medicines Generic medicines Percentage
Without alternative With alternative
Graphic 4 – Existence of therapeutic alternatives in generic and non-generic medicines
Table II – Codification of quality alerts publicly available from the Portuguese National Authority of Medicines and Health Products (data from January 2003 to 13th of June 2010)
Code Frequency (n=240) Percentage Reason Specifications F 64 26,7 Primary packaging A 39 16,3 Labelling P 31 12,9 Stability G 22 9,2 Package leaflet I 17 7,1 Packaging E 15 6,3 Formula K 14 5,8 Device D 9 3,8 Contamination B 6 2,5 Package leaflet and labelling J 6 2,5 Regulatory O 6 2,5 Manufacture H 5 2,1 GMP L 2 0,8 Counterfeiting C 1 0,4 Information for health professionals M 1 0,4 No reason stated N 1 0,4 Validation of analytical methods Q 1 0,4
Number of alerts within each reason for quality alert 64 39 31 22 17 15 14 9 6 6 6 5 2 1 1 1 1 0 10 20 30 40 50 60 70 F A P G I E K D B J O H L C M N Q Reason Number of quality alerts F: Specifications A: Primary packaging P: Labelling G: Stability I: Package leaflet
J: Package leaflet and labelling
Graphic 5 – Pareto analysis of classes of quality alerts, as identified in Table II
A fourth major group was composed of problems with information to medicine users (classes P, I and J). A Pareto analysis (Graphic 5) shows the contribu-tion of these three main reasons and of the fourth rea-son composed of the classifications Labelling, Pack-age Leaflet, PackPack-age Leaflet and Labelling.
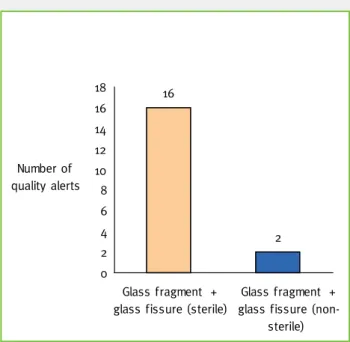
A further analysis into the problems grouped un-der Primary Packaging revealed that 46,2% of these quality alerts were associated with the use of glass (Graphic 6). Most of the medicines within these quality alerts were sterile medicines (Graphic 7). Thus consciousness of the higher risk that seems to be associated with use of glass in primary packaging, is strengthened when it is realised the impact as this is higher with sterile than with non-sterile medicines. Impact from a quality alert could also come from out of stock of medicines due to the quality problem. As with the out of stock medicines list, analysis of the
existence of therapeutic alternatives for medicines with quality alerts showed that impact, and thus risk, is lower for generic than for non-generic medicines. Finally, a classification system adopted for the safety alerts is presented in Table III.
Graphic 6 – Reasons for quality alerts due to primary packaging
7,7 5,1 5,1 2,6 2,6 2,6 2,6 2,6 2,6 2,6 2,6 2,6 2,6 2,6 2,6 2,6 2,6 2,6 2,6 2,6 38,5 0 5 10 15 20 25 30 35 40 45 G la ss f ra gme nt G la ss f is sur e Cl os ur e tig htn ess No n-c or form ity of p ac ka gin g mater ia l In k p ar tic le s Pe rfor at ed bl ist er Te flo n p art ic le s in s ol ut io n St rang e b od y St op pe r pa rt ic le s Tu be in ne r co at Ne ed le s he ath Mi ss ing mar k for s us pe nsi on r ec on st itut io n Tw o ta ble ts in sa m e b list er a lv eo lu s So lu tio n depo si t ou ts id e amp ou le D efor m ed c ap su le Pa ck ag in g c lo su re De fe ct d uri ng p ac ka gin g m an ufa ct ur e Ru bb er plun ge r Ro ug hn es s of bl ist er s ealing Seal ing Fin ge r su pp ort of s yri ng e de ta ch ed Percentage 16 2 0 2 4 6 8 10 12 14 16 18 Glass fragment + glass fissure (sterile)
Glass fragment + glass fissure
(non-sterile) Number of
quality alerts
Graphic 7 – Quality alerts due to primary packaging in sterile and non-sterile medicines
The majority of safety alerts were derived from evalu-ation of the risk-benefit balance of medicines and risk communication to health professionals and general public. Quality issues were responsible for only 5,4% of safety alerts. As concerns availability of medicines, marketing authorizations remained valid in more than 80% of the cases (Table IV). In this regard impact of safety alerts is more related to
re-latter ones if a problem with the alternatives were also detected.
Conclusion
The use of risk management is considered useful both in the design of regulations and in their implementa-tion by regulatory bodies.
Governments have promoted use of risk to develop a better regulation environment, more efficiency in government services and reduce costs for business-es. A drive for the development of a policy based on risk was the reduction of burdensome on business to improve competitiveness. Better resource allocation should benefit businesses, as with regulator bodies. Use of risk as a regulatory tool has been linked to ad-vancements in businesses, processes, products and
outcomes understanding. The increase in knowledge
has allowed going from a deterministic regulation to a more risk-based one in the nuclear and pharmaceu-tical industries.
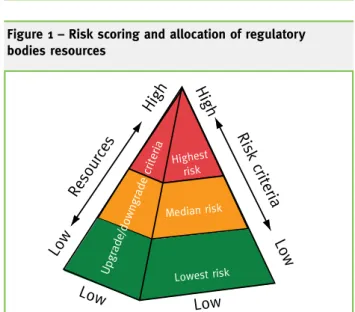
Regulator bodies of distinct areas have introduced risk as an element to structure their activities in delivering regulation. The drivers for the use of risk were a better allocation of resources in response to increasing demands on regulators, by using commen-surate regulatory responses according to risk (as in risk-based inspections and differentiated licensing); encourage and facilitate technological development and continuous improvement (as exemplified by strictions in the use of medicines. Use of the information from a risk analysis allows to score risk and allocate resources (Figure 1): the pyramid has been divided into three levels – three risk scores, be-ing higher risk at the top related to a higher allocation of resources. The front face determines risk cri-teria defining where to place firms, sites, products or processes. On the side face other criteria may define that in particular cases risk scores are to be upgraded or
downgrad-ed, e.g. to biase decision making in
managing high institutional risks. In our simple example generic and non-generic medicines without therapeutic alternatives could be at the top of the pyramid, while ones with therapeutic alternatives would be placed in the middle. A side criterion would upgrade the
Table III – Codification of safety alerts publicly available from the Portuguese National Authority of Medicines and Health Products (data from July 2002 to 13th of June 2010)
Frequency (n=149) Percentage Reason Risk-benefit balance 121 81,2 Quality 8 5,4 Pandemic Flue 5 3,4 Safety variation 4 2,7 Safety concern 3 2 SPC and PIL not updated with safety information 1 0,7 Interaction 1 0,7 Safety information to be included in PIL 1 0,7 Pharmacovigilance notification 1 0,7 Clinical information 1 0,7 Mode of use 1 0,7 Information for the public and heath professionals 1 0,7 Information for the public 1 0,7
Table IV – Status of the Marketing Authorization (MA) in result of the safety alert
Frequency (n=149) Percentage MA status Valid 122 81,9 Suspended 13 8,7 Revoked 7 4,7 Recall 6 4 Conditional 1 0,7
Figure 1 – Risk scoring and allocation of regulatory bodies resources
risk-based review systems); increase safety (e.g. of
medicines through use of risk management plans) and quality, and the use of risk as a tool to help in decision-making.
It was recognised that limited resources were being equally spent in activities with different risks, and a shift was needed towards allocation of resources to activities that posed greater risks. Through the use of risk it is possible to achieve better results using the same resources. Overall, regulatory scrutiny should adjust to the degree of risk associated with processes, products and outcomes.
Through the analysis of an out of stock medicines list, quality alerts and safety alerts it has been seen that information is available for regulator bodies to use risk as an element to structure their activities in implementing regulation. A simple improvement of the analysis made could be achieved if data on manufacturers and actual availability of the medicine in the market were introduced in the analysis. Cer-tainly within a regulator body other data and knowl-edge can be used to develop a risk-based framework within the organization.
In conclusion risk seems a valid regulatory tool to increase effectiveness through better organization of processes and management of regulatory resources and in this way improve results of regulatory activity without putting in jeopardy their quality.
Low High Reso urce s High Low Ris k c rite ria Highest risk Median risk Lowest risk Low Low Upgrade/downgrade criteria
References
1 Ahn SK, Kim SI, Oh KM. Deterministic and risk-informed ap-proaches for safety analysis of advanced reactors: Part I, deter-ministic approaches. Reliability Engineering and System Safety. 2010;95:451-458.
2 Bartle I. Risk-based regulation and better regulation in the UK: towards what model of risk regulation? Paper prepared for presen-tation at the 2nd Biennial Conference of the ECPR Standing Group on Regulatory Governance, Utrecht University, the Netherlands. ‘(Re)Regulation in the Wake of Neoliberalism. Consequences of Three Decades of Privatization and Market Liberalization’; June 5th-7th 2008.
3 Flueler T, Seiler H. Risk-based regulation: a suitable concept to legislate and regulate technical risks? Evaluation of various case studies in Switzerland, in: Goossens, L.H.J. (Ed.), Proceedings 9th annual conference on risk analysis: facing the new millennium. Delft: Delft University Press; 1999. p. 202-206.
4 Gary B, et al. Rethinking regulation: report of the taskforce on re-ducing regulatory burdens on business. Report to the Prime Min-ister and the Treasurer (Australian government), Canberra. 2006. 5 European Commission (EC). Impact assessment guidelines (SEC(2009) 92). 2009.
6 European Commission (EC) better regulation [homepage on the
internet]. [Updated 2010 April 23; cited 2010 May 6]. Available from: http://ec.europa.eu/governance/better_regulation/index_en.htm. 7 Hampton P. Reducing administrative burdens: effective inspec-tion and enforcement. HM Treasury. 2005.
8 Christiane Arndt C et al. Indicators of regulatory management systems. Regulatory policy committee, 2009 report, OECD. 2009. 9 Connor JA, McHugh TE. Impact of risk-based corrective ac-tion (RBCA) on State LUST Corrective Acac-tion Programs. Hu-man and Ecological Risk Assessment: An International Journal. 2002;8(3):573-589.
10 Environment Agency UK. Delivering for the environment, a 21st approach to regulation. Environment Agency UK. 2005.
11 Environment Agency UK. Operational risk appraisal (Opra). [Inter-net page]. [updated 2010 April 23; cited 2010 May 30]. Available from: www.environment-agency.gov.uk/business/regulation/31827.aspx. 12 Environment and Heritage Service. Better regulation for a better environment. Environment & Heritage Service. 2008.
13 ERMA. Approach to risk, position paper on the approach to risk, methodologies for dealing with this and the technical and commu-nity information required for implementation. Environmental Risk Management Authority (ERMA) (New Zealand). 2002.
14 ERMA. Environmental Risk Management Authority (ERMA) (New Zealand) [homepage on the internet]. [cited 2010 May 6]. Available from: www.ermanz.govt.nz/.
15 Johnstone D. SEPA Risk Assessment. Scottish Environmental Protection Agency. 2005.
16 Marsden M, Harley D. Regulatory risk assessment processes. Scottish Environment Protection Agency. 2005.
17 Pollard SJ, et al. Current Directions in the Practice of Environ-mental Risk Assessment in the United Kingdom. Environ. Sci. Technol. 2002;36:530-538.
18 United States Environmental Protection Agency. Study results and recommendations for risk-based decision-making program performance monitoring, risk-based decision making performance assessment study bulletin #2. United States Environmental Protec-tion Agency. 2002.
19 Water Environment (Controlled Activities) (Scotland) Regula-tions, 2005. No 348.
20 FDA. Part 11, electronic records; electronic signatures - scope and application. U.S. Department of Health and Human Services Food and Drug Administration. 2003.
21 FDA. Sterile drug products produced by aseptic processing – cur-rent good manufacturing practice. U.S. Department of Health and Human Services Food and Drug Administration. 2004.
22 FDA. Pharmaceutical cGMPS for the 21st century – a risk-based approach final report. Department of Health and Human Services U.S Food and Drug Administration. 2004.
23 FDA. Risk-based method for prioritizing cGMP inspections of pharmaceutical manufacturing sites – a pilot risk ranking model. Department of Health and Human Services U.S. Food and Drug Ad-ministration. 2004.
24 FDA. Pharmaceutical quality for the 21st century a risk-based ap-proach progress report. Department of Health and Human Services
U.S. Food and Drug Administration [internet]. 2007 May [cited 2010 March 27]. Available from: www.fda.gov/AboutFDA/Center-sOffices/CDER/ucm128080.htm.
25 FDA. Pharmaceutical cGMPS for the 21st century – a risk-based approach: second progress report and implementation plan. Food and Drug Administration. [internet]. 2009 August 17 [cited 2010 May 6]. Available from: www.fda.gov/Drugs/DevelopmentAp- provalProcess/Manufacturing/QuestionsandAnswersonCurrent-GoodManufacturingPracticescGMPforDrugs/UCM071836. 26 FDA. Food and Drug Administration compliance program guidance manual – chapter 46 – new drug evaluation (program 7346.832). Food and Drug Administration. 2010.
27 MHRA. Good Clinical Practice: Risk-based inspections. MHRA [Internet page]. [updated 2010 March 25; cited 2010 April 29].
Avail-able from: www.mhra.gov.uk/PrintPreview/DefaultSplashPP/
CON046603?.
28 Rönninger IS, Schmitt S. Health authority special report: MHRA’s risk-based inspections seminar. Parenteral Drug As-sociation [homepage on the Internet]. [cited 2010 March 28]. Available from: www.pda.org/MainMenuCategory/QualityRegu- latoryAffairs/Health-Authority-Special-Report-MHRAs-Risk-Based-Inspections-Seminar.aspx.
29 Wechsler J. FDA seeks risk-based inspection program. Pharma-ceutical Technology. 2003:26-29.
30 EFPIA. Good Manufacturing Practices (GMP) – inspections of global pharmaceutical supply chains. EFPIA scientific, technical & regulatory affairs publications. 2009.
31 Commission Regulation (EC) No 1234/2008, 2008. Commission Regulation (EC) No 1234/2008 of 24 November 2008. Official Jour-nal L 334, 12/12/2008. p. 7-24.
32 European Commission (EC). Communication from the Commis-sion – guideline on the details of the various categories of variations to the terms of marketing authorisations for medicinal products for human use and veterinary medicinal products (2010/C 17/01) of 22 January 2010. European Commission (EC), eudraLex – volume 2 – pharmaceutical legislation -notice to applicants and regulatory guidelines medicinal products for human use.
33 FDA. Comparability protocols – protein drug products and bio-logical products – chemistry, manufacturing, and controls Informa-tion (draft guidance). U.S. Department of Health and Human Ser-vices Food and Drug Administration (FDA). 2003.
34 FDA. Comparability protocols – chemistry, manufacturing, and controls information (draft guidance). U.S. Department of Health and Human Services Food and Drug Administration. 2003. 35 FDA. Question-based review (QbR) for generic drugs: an en-hanced pharmaceutical quality assessment system food and drug administration. Food and Drug Administration, Center for Drug Evaluation and Research, Office of Generic Drugs [internet]. [up-dated 2009 April 30; cited 2010 March 26]. Available from: www. fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevel- opedandApproved/ApprovalApplications/AbbreviatedNewDru-gApplicationANDAGenerics/ucm120973.htm.
36 Yu L. Pharmaceutical quality by design: product and process de-velopment, understanding, and control. Pharmaceutical Research. 2008;25(4).
37 Yu L et al. US FDA question-based review for generic drugs: a new pharmaceutical quality assessment system. 2007;4(4):239-248. 38 Cogdill RP, Drennen JK. Risk-based quality by design (QbD): a Taguchi perspective on the assessment of product quality, and the quantitative linkage of drug product parameters and clinical perfor-mance. J Pharm Innov. 2008;3:23-29.
39 Garcia T, Cook G, Nosal R. PQLI key topics - criticality, design space, and control strategy. J Pharm Innov. 2008;3:60-68.
40 Lepore J, Spavins J. PQLI design space. J Pharm Innov. 2008;3:79-87. 41 Lionberger AR, et al. Quality by design: concepts for ANDAs. The AAPS Journal. 2008;10(2).
42 ICH. Pharmaceutical development Q8(R2). International con-ference on harmonisation of technical requirements for registration of pharmaceuticals for human use (ICH). 2009.
43 ICH. Quality risk management Q9. International conference on harmonisation of technical requirements for registration of pharma-ceuticals for human use (ICH). 2005.
44 ICH. Pharmaceutical quality system Q10. International confer-ence on harmonisation of technical requirements for registration of pharmaceuticals for human use (ICH). 2008.
45 Kadac AC, Matsuo T. The nuclear industry’s transition to risk-informed regulation and operation in the United States. Reliability Engineering and System Safety. 2007;92:609-618.
46 Modarres M. Advanced nuclear power plant regulation using risk-informed and performance-based methods. Reliability Engi-neering and System Safety. 2009;94:211-217.
47 Jamali K. Use of risk measures in design and licensing of future re-actors. Reliability Engineering and System Safety. 2010;95:935-943. 48 Kim SI, Ahn SK, Oh KM. Deterministic and risk-informed ap-proaches for safety analysis of advanced reactors: Part II, Risk-informed approaches. Reliability Engineering and System Safety. 2010;95:459-468.
49 Mishra A, Pandey MD, Chauhan A. Regulation of nuclear pow-er plants a multi objective approach. Annals of Nuclear Enpow-ergy. 2009;36:1560-1573.
50 Stärk DCK, et al. Concepts for risk-based surveillance in the field of veterinary medicine and veterinary public health: Review of cur-rent approaches. BMC Health Serv Research. 2006;6:20.
51 EMA, HMA. Action plan to further progress the European risk management strategy. European Medicines Agency (EMA). 2005. 52 EMA, HMA. Implementation of the action plan to further progress the European risk management strategy: rolling two-year work pro-gramme (2008-2009). European Medicines Agency (EMA). 2007. 53 EMA, HMA. European risk management strategy: 2008-2009 work programme adopted. European Medicines Agency (EMA). 2008. 54 EMA, HMA. 2008 Public status report on the implementation of the European risk management strategy. European Medicines Agency (EMA). 2009.
55 Directive 2001/83/EC, 2001. Directive 2001/83/EC of the Euro-pean Parliament and of the Council of 6 November 2001. Official Journal L 106, 17/4/2001, 1-39.
56 European Commission (EC). Volume 9A of the rules governing medicinal products in the European Union – guidelines on pharma-covigilance for medicinal products for human use. European Com-mission (EC), eudraLex – volume 9 pharmacovigilance guidelines. 57 EMA. Guideline on risk management systems for medicinal prod-ucts for human use. European Medicines Agency (EMA). 2005. 58 Directive 2010/84/EU, 2010. Directive 2010/84/EU of the Euro-pean Parliament and of the Council of 15 December 2010. Official Journal L 348, 31/12/2010, 74-99.
59 Regulation (EU) No 1235/2010 of the European Parliament and of the Council of 15 December 2010. Official Journal L 348, 31/12/2010, 1-16. 60 Bounds G. Achieving better quality regulations - incorporating risk assessment tools in RIA to prepare better rules. OECD [inter-net]. 2009 November 24 [cited 2010 April 1]. Available from: www. oecd.org/dataoecd/31/4/44844265.pdf.
61 Gregory B et al. Regulatory impact analysis: a tool for policy co-herence. OECD browse_it editions. 2009.
62 European Environment Agency. Environmental Risk Assess-ment – Approaches, Experiences and Information Sources. Europe-an Environment Agency [internet]. Environmental issue report No 4; 1998 March 24 [cited 2010 April 1]. Available from: http://www. eea.europa.eu/publications/GH-07-97-595-EN-C2.
63 Rothstein H et al. The risks of risk-based regulation: insights from the environmental policy domain. Environment International. 2006;32:1056-1065.
64 Black J. Risk-based regulation, choices, practices and lessons be-ing learnt, in: Bounds, G., et al. (Eds.), Risk and regulatory policy: improving the governance of risk. OECD browse_it editions. 2010. 65 INFARMED, I.P.. Circular Informativa n.º 024/CA – ruptura de fornecimento de medicamentos. Autoridade Nacional do Medica-mento e Produtos de Saúde, I.P. (INFARMED, I.P). 2005.
66 INFARMED, I.P.. Deliberação n.º 108/CA/2005. Autoridade Na-cional do Medicamento e Produtos de Saúde, I.P. (INFARMED, I.P). 2005.
67 INFARMED, I.P.. Autoridade Nacional do Medicamento e Produ-tos de Saúde, I.P. (INFARMED, I.P), ruptura de medicamenProdu-tos. [Homepage on the internet]. Available from: http://app.infarmed. pt/sgrt/listrstock.aspx.
68 INFARMED, I.P.. Autoridade Nacional do Medicamento e Produtos de Saúde, I.P. (INFARMED, I.P), alertas [Homepage on the internet]. Available from: www.infarmed.pt/portal/page/portal/ INFARMED?categoria=CAT_ALERTAS.
69 APOGEN. Mercado de Medicamentos Genéricos. Associação Portuguesa de Medicamentos Genéricos (APOGEN). [internet]. [cited 2010 May 30]. Available from: http://www.apogen.pt/con-teudos/SystemPages/page.asp?art_id=50.