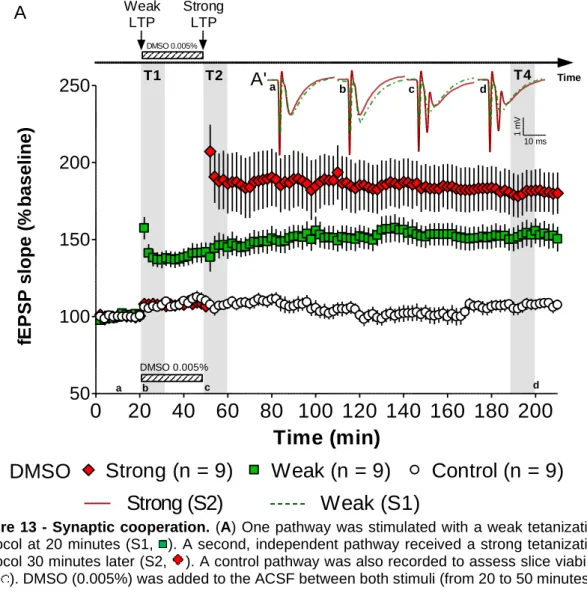
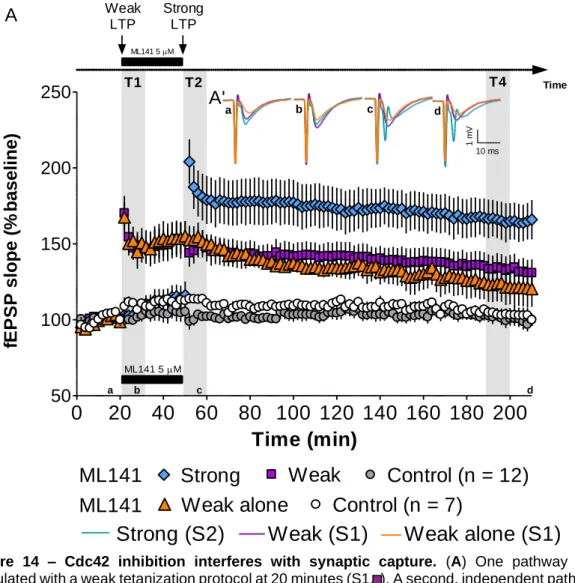
The role of Cdc42 GTPase during synaptic cooperation
Texto
Imagem




Documentos relacionados
The probability of attending school four our group of interest in this region increased by 6.5 percentage points after the expansion of the Bolsa Família program in 2007 and
Foi então que para mim surgiram os primeiros obstáculos de muitos outros que ainda estavam por vir; primeiro eu era uma das pessoas excluída do mundo digital com um agravante de
Suínos de dez lotes encaminhados para um abatedouro-frigorífico legalmente estabelecido, cadastrado e inspecionado pela Secretaria da Agricultura, Pecuária e Irrigação do Rio
Além disso, o Facebook também disponibiliza várias ferramentas exclusivas como a criação de eventos, de publici- dade, fornece aos seus utilizadores milhares de jogos que podem
H„ autores que preferem excluir dos estudos de prevalˆncia lesŽes associadas a dentes restaurados para evitar confus‚o de diagn€stico com lesŽes de
Ousasse apontar algumas hipóteses para a solução desse problema público a partir do exposto dos autores usados como base para fundamentação teórica, da análise dos dados
didático e resolva as listas de exercícios (disponíveis no Classroom) referentes às obras de Carlos Drummond de Andrade, João Guimarães Rosa, Machado de Assis,
Apesar destes não virem expressamente previstos na CEDH, o art.º 2.º, do Protocolo I, refere-se ao direito à educação, e o TEDH já se pronunciou sobre o direito à saúde, o
