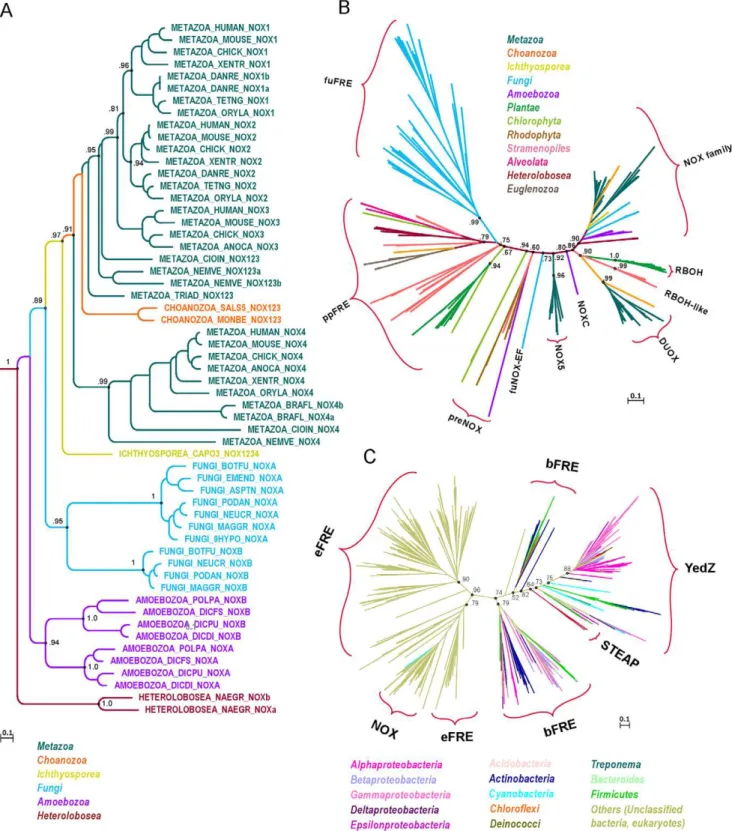
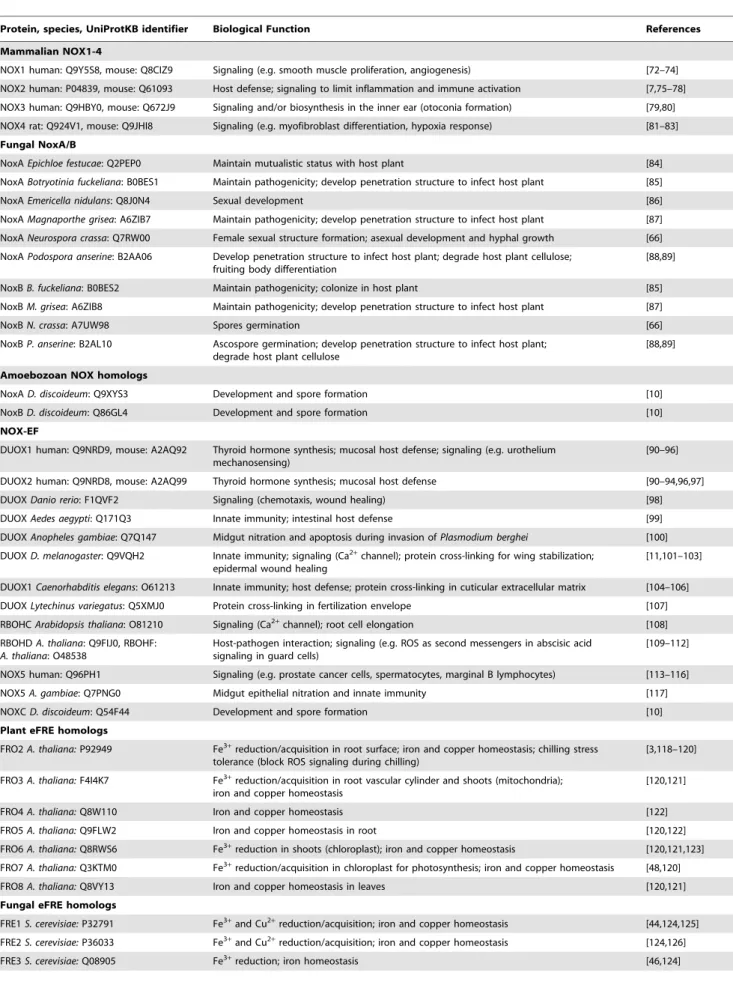
Evolution of the ferric reductase domain (FRD) superfamily: modularity, functional diversification, and signature motifs.
Texto
Imagem

Documentos relacionados
Ao Dr Oliver Duenisch pelos contatos feitos e orientação de língua estrangeira Ao Dr Agenor Maccari pela ajuda na viabilização da área do experimento de campo Ao Dr Rudi Arno
Ousasse apontar algumas hipóteses para a solução desse problema público a partir do exposto dos autores usados como base para fundamentação teórica, da análise dos dados
The fourth generation of sinkholes is connected with the older Đulin ponor-Medvedica cave system and collects the water which appears deeper in the cave as permanent
In the hinterland (Lika region) they are partly permeable but on higher positions at Velebit Mt. calcareous breccias are highly permeable. The best prove for the mentioned is
didático e resolva as listas de exercícios (disponíveis no Classroom) referentes às obras de Carlos Drummond de Andrade, João Guimarães Rosa, Machado de Assis,
i) A condutividade da matriz vítrea diminui com o aumento do tempo de tratamento térmico (Fig.. 241 pequena quantidade de cristais existentes na amostra já provoca um efeito
A aula que vou apresentar, com um plano meu em anexo (Anexo F), corresponde ao plano 14 da unidade 3do manual, disponível em anexo (Anexo G), e foi das poucas aulas em
Isto é, o multilateralismo, em nível regional, só pode ser construído a partir de uma agenda dos países latino-americanos que leve em consideração os problemas (mas não as
