Flávia da Silva Nader Motta
149
0
0
Texto
Imagem
+7
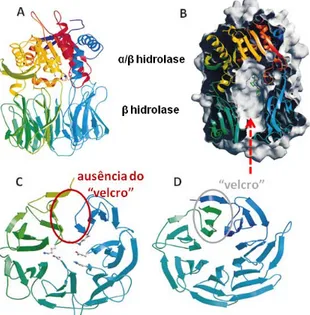
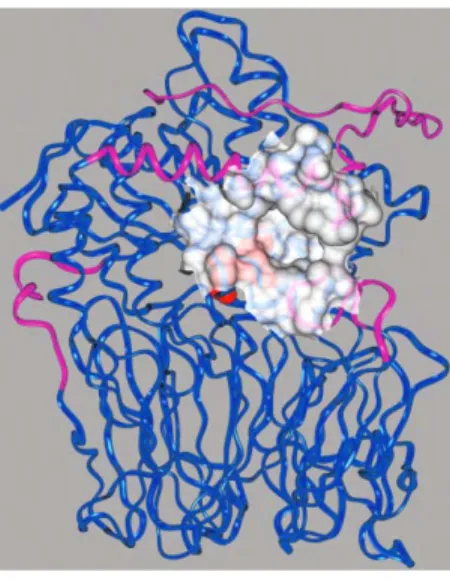
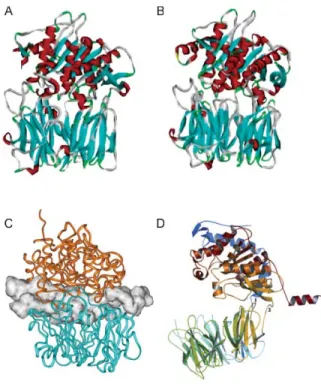
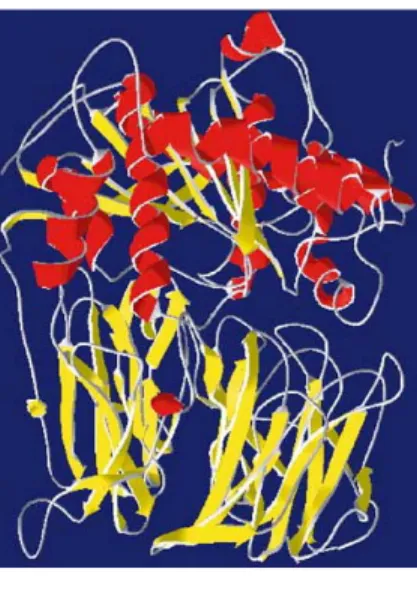
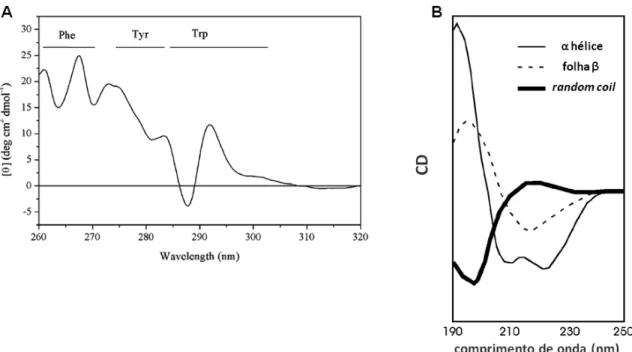
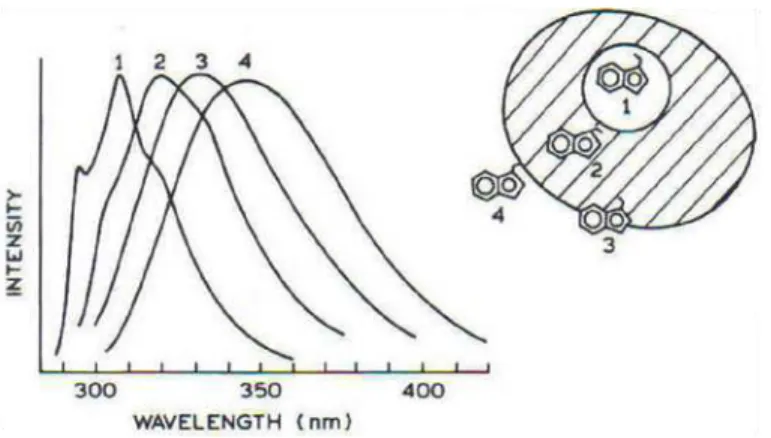
Documentos relacionados