w w w . r e u m a t o l o g i a . c o m . b r
REVISTA
BRASILEIRA
DE
REUMATOLOGIA
Review
article
Primary
immunodeficiency
association
with
systemic
lupus
erythematosus:
review
of
literature
and
lessons
learned
by
the
Rheumatology
Division
of
a
tertiary
university
hospital
at
São
Paulo,
Brazil
Paolo
Ruggero
Errante
a,b,
Sandro
Félix
Perazzio
b,
Josias
Brito
Frazão
a,
Neusa
Pereira
da
Silva
b,
Luis
Eduardo
Coelho
Andrade
b,∗aDepartmentofImmunology,InstituteofBiomedicalSciences,UniversidadedeSãoPaulo(USP),SãoPaulo,SP,Brazil bDepartmentofMedicine,UniversidadeFederaldeSãoPaulo(UNIFESP),SãoPaulo,SP,Brazil
a
r
t
i
c
l
e
i
n
f
o
Articlehistory:
Received12March2014 Accepted8March2015
Availableonline1September2015
Keywords:
Autoimmunedisease Primaryimmunodeficiency Systemiclupuserythematosus Antibodiesdeficiency
a
b
s
t
r
a
c
t
Primaryimmunodeficiencydisorders(PID)representaheterogeneous groupofdiseases resultingfrominheriteddefectsinthedevelopment,maturationandnormalfunctionof immunecells;thus,turningindividualssusceptibletorecurrentinfections,allergy, autoim-munity,and malignancies.In this retrospectivestudy,autoimmune diseases (AIDs), in specialsystemiclupuserythematosus(SLE)whicharoseassociatedtothecourseofPID, aredescribed.Classically,theliteraturedescribesthreegroupsofPIDassociatedwithSLE: (1)deficiencyofComplementpathwaycomponents,(2)defectsinimmunoglobulin synthe-sis,and(3)chronicgranulomatousdisease(CGD).Currently,otherPIDhavebeendescribed withclinicalmanifestationofSLE,suchasWiskott–Aldrichsyndrome(WAS),autoimmune polyendocrinopathycandidiasisectodermaldystrophy(APECED),autoimmune lymphopro-liferativesyndrome(ALPS)andidiopathicCD4+lymphocytopenia.Alsowepresentfindings
fromanadultcohortfromtheoutpatientclinicoftheRheumatologyDivisionof Universi-dadeFederaldeSãoPaulo.ThePIDmanifestationsfoundbyourstudygroupwereconsidered mildintermsofseverityofinfectionsandmortalityinearlylife.Thus,itispossiblethatsome immunodeficiencystatesarecompatiblewithsurvivalregardinginfectioussusceptibility; howeverthesestatesmightrepresentastrongpredisposingfactorforthedevelopmentof immunedisorderslikethoseobservedinSLE.
©2015ElsevierEditoraLtda.Allrightsreserved.
∗ Correspondingauthor.
E-mail:[email protected](L.E.C.Andrade). http://dx.doi.org/10.1016/j.rbre.2015.07.006
Associac¸ão
de
imunodeficiência
primária
com
lúpus
eritematoso
sistêmico:
revisão
da
literatura
e
as
lic¸ões
aprendidas
pela
Divisão
de
Reumatologia
de
um
hospital
universitário
terciário
em
São
Paulo
Palavras-chave: Doenc¸aautoimune Imunodeficiênciaprimária Lúpuseritematososistêmico Deficiênciadeanticorpos
r
e
s
u
m
o
Asimunodeficiênciasprimárias(IDP)representamumgrupoheterogêneodedoenc¸as resul-tantesdedefeitoshereditáriosnodesenvolvimento,maturac¸ãoefunc¸ãonormaldecélulas dosistemaimunológico;assim,tornamosindivíduossuscetíveisainfecc¸õesrecorrentes, alergia, autoimunidade e doenc¸as malignas. Neste estudo retrospectivo, descrevem-se doenc¸asautoimunes(DAI),emespecialolúpuseritematososistêmico(LES),quesurgiram associadasaocursodasIDP.Classicamente,aliteraturadescrevetrêsgruposdeIDP associ-adasaoLES:(1)deficiênciadecomponentesdaviadocomplemento,(2)defeitosnasíntese deimunoglobulinase(3)doenc¸agranulomatosacrônica(DGC).Naatualidade,outrasIDP têmsidodescritascomomanifestac¸õesclínicasdoLES,comoasíndromedeWiskott–Aldrich (WAS),apoliendocrinopatiaautoimune-candidíase-distrofiaectodérmica(APECED),a sín-dromelinfoproliferativaautoimune(ALPS)ealinfocitopeniaidiopáticaCD4+.Tambémsão
apresentadosachadosdeumacoortedeadultosdoambulatóriodaDivisãode Reumatolo-giadaUniversidadeFederaldeSãoPaulo.Asmanifestac¸õesdeIDPencontradaspelonosso grupodeestudoforamconsideradaslevesemtermosdegravidadedeinfecc¸õese mortal-idadenoiníciodavida.Assim,épossívelquealgunsestadosdeimunodeficiênciasejam compatíveiscomasobrevivênciaemrelac¸ãoàsuscetibilidadeinfecciosa;noentanto,estes estadospodemrepresentarumfatordepredisposic¸ãoforteparaodesenvolvimentode doenc¸asimunológicas,comoobservadonoLES.
©2015ElsevierEditoraLtda.Todososdireitosreservados.
Introduction
Primaryimmunodeficiencydisorders(PID)representa hetero-geneousgroupofdiseasesresultingfrominheriteddefectsin thedevelopment,maturationandnormalfunctionofimmune cells. PID often have an important genetic basis leading to different immune disorders associated with infections, autoimmunediseases and other malignanciesinpatients.1 Since these are congenital conditions, usually with well-definedgeneticdefectsandmendelianinheritance,children are the most predominant patients. On the other hand, autoimmunediseases (AIDs) haveacomplex multifactorial polygenicetiology inwhich environmentaltriggersplayan importantrole intheir pathogenesisand representagroup ofmorethan70knowndiseases.2Remarkably,AIDsrepresent oneofthemostcommonclinicalphenotypesofmanyforms ofPID,onlyovercomebythefrequencyofinfections.3
Systemic lupus erythematosus (SLE) is a multi-organ autoimmune disease characterized by a range of clinical manifestationsthatpredominantlyaffectswomenin repro-ductiveage.InSLE,polyclonalhypergammaglobulinemiaand multipleautoantibodiesareproducedpredominantlyagainst nuclear antigens. These autoantibodies deposit on several organs, including kidneys, skin and joints, causing severe inflammation.4 Although SLE patients have hypergamma-globulinemia,theyoftenpresentsevereinfections,especially whilereceivingimmunosuppressivetreatment.
Infections by opportunistic pathogens are commonly seen in patients with PID.5 These infections, either
clinical or subclinical, may represent the primary trigger for the development of autoimmunity. In genetically pre-disposed individuals, chronic exposure to environmental factors can promote the development of autoantibodies many years before the disease onset. Patients with SLE present an increased susceptibility to infection in pre-clinicalphaseofdisease.6Classically,theliteraturedescribes three groups of PID associated with SLE: (1) deficiency of Complement pathway components7; (2) selective and partial defects in immunoglobulin synthesis (particularly isolated IgA and IgM deficiencies)8,9; and (3) chronic gran-ulomatous disease (CGD).9–12 However, among clinical observations, several other PID may also occasionally be associated with SLE or SLE-like syndrome manifestations. These include Wiskott–Aldrich syndrome (WAS),13 autoim-munepolyendocrinopathycandidiasisectodermaldystrophy (APECED),14 autoimmune lymphoproliferative syndrome (ALPS),15 idiopathic CD4+ lymphocytopenia (ICL),16 partial T cell immunodeficiency and hyper-immune dysregula-tion (including autoimmunity, inflammatory diseases and elevatedIgEproduction).17
Classical
and
non-classical
PID
associated
with
SLE
PID are a heterogeneous group of diseases characterized byincreasedsusceptibilityto multipleandrecurrent infec-tions causedbyvirulent and non-virulent microorganisms. TheliteratureranksPIDinclassicalandnon-classicalforms. ClassicalPIDaredefinedonthebasisofanovert immuno-logic phenotype, often leading to the identification of the disease-causing gene. The expert committee on Primary Immunodeficiency of the International Union of Immuno-logicalSocieties(IUIS)recentlyupdatedthe classificationof humanclassicalPID.19Non-classicalPIDaredefinedonthe basisofaspecificthoughunremarkableclinicalphenotype, andneverhavebeenclassifiedasafullydistinctphenotypeout ofPIDclassification.Additionally,theyhavenotbeenincluded intheupdatedclassificationofPID,compiledbytheadhoc ExpertCommitteeoftheIUIS.20However,thefactthat non-classicalPIDmaynotbeassociatedwithrecurrentinfections does not guarantee that these diseases do not predispose thedevelopmentofautoimmunedisorders.Therefore,inthis paper,wedescribethemajorclassicalandnon-classicalPID associatedtoSLE.
Complementdeficiencies
TheComplementsystemiscomposedbyagroupofplasma and surface cell-proteins with important role in innate andacquired humoralimmunesystem, responsibleforthe destructionofmicrobialagentsandclearanceofcirculating immunecomplexes.21InSLE,thedepositionofimmune com-plexescontainingmultipleautoantibodiesandactivationof theComplement systemmediatetissuedamage.4 Paradox-ically, deficienciesin components ofearly elements ofthe classicalpathway(C1q,C1r,C1s,C4,andC2)arestrongly asso-ciatedwiththedevelopmentofSLE.Inaddition,deficiency incomponentsofthelatecommonpathway(C5,C6,C7,C8a andC8b)aswellassomeelementsofthealternative path-way(C3andFactorI)areonlyoccasionallyassociatedwithSLE (Table1).19 Geneticdeficienciesofthesecomponentsmight contributetowardsSLEpathogenesisbydecreasingimmune complex clearance capacity. The literature is controversial inrespecttomannose-bindinglectin(MBL)22,23and antibod-iesagainstMBL24inthepathogenesisofSLE.Someauthors, andthisincludesourgroup,havedescribedthepresenceof increasedMBLdeficiencyinSLEpatients(unpublisheddata). However,furtherstudiesshouldbeconductedforabetter elu-cidationofthisassociationwithSLE.
Defectsinimmunoglobulinsynthesis(antibody deficiencies)
Antibodydeficienciesalsoreferredasimmunoglobulin defi-ciencies,representagroupofdiseases(immunesystem disor-ders)characterizedbyloworabsentlevelsofimmunoglobulin inthe blood. Immunoglobulins (Ig) are largey-shaped gly-coprotein molecules produced byB cells that detect, bind and neutralize foreign substances (like bacteria, viruses, fungi, toxins and allergens). They also have the capability
tosignalimmunecellstoeliminateforeignsubstances. Anti-body deficiencies represent a group of diseases and are consideredthemostcommontypeofprimaryimmune defi-cienciesinhumans.Duetothefactthatprotectivelevelsof IgG that are passively acquired bythe newbornsfrom the mother decreasesduringthefirstyearoflife,symptomsof thisgroupofdiseasesonlybecomesymptomaticattheend ofthefirstyearoflife.Thespectrumofantibodydeficiencies isbroad,rangingfromtheabsenceofBcellsandserumIgs (mostseveretypeofantibodydeficiency)toselectiveantibody deficiencywithnormalserumlevelsoftotalimmunoglobulin. Inaddition toincreasedsusceptibilitytoinfections,clinical presentationofantibodydeficienciesmayalsoincludeother diseaseprocesses(e.g.,autoimmunityandmalignancies).
Commonvariableimmunodeficiencydisorders
Common variable immunodeficiency (CVID) is a heteroge-neous group ofprimaryantibodydeficiencies diagnosedin humans,withbroadclinicalspectrum.CVIDpatientspresent history of hypogammaglobulinemia, recurrent respiratory tractinfections,buttheclinicalspectrummayinclude autoim-munephenomena,bowelinflammatoryorinfectiousdisease, and granulomatous disease which can affect liver, spleen and lungs.25 Ithasbeen postulated thatpersistentantigen stimulation,recurrenttissuedamage,defectiveclearanceof immune complexes and immune dysregulation contribute towardthedevelopmentofautoimmunity,includingSLE,but more frequently autoimmune cytopenia and endocrinopa-thy.Fernandez-Castroetal.describedaseriesof18patients with SLE and CVID. Interestingly, up to 67% of them had theautoimmunediseasecontrolledafterthedevelopmentof theimmunodeficiency.26Geneticabnormalitiesdescribedin CVID includedefects inthe inducible co-stimulator(ICOS), the membrane activator and calcium-modulator interactor (TACI), the B-cellactivating factor receptor (BAFF-R),CD19, CD20andCD81(Table2).19AlthoughCVIDhasbeendescribed inpatientsafterthediagnosisofSLE,26,27 immunosuppress-iveagentsusedforSLEtreatmentcanbetheverycauseof hypogammaglobulinemiadevelopment,turningthedefinitive diagnosis ofCVID into a difficulttask, sincethe diagnosis ofCVIDdependsonexclusionofallotherknowncausesof hypogammaglobulinemia.
SelectiveIgAdeficiency
Table1–ComplementdeficienciesassociatedwithSLEorSLE-likemanifestation.
Disease Associationfeaturesandautoimmunemanifestation Inheritance Defectivegene OMIMnumber
C1qdeficiency SLE-likesyndrome,rheumatoidsyndrome, infections
AR C1QA 120550
C1QB 601269
C1QC 120575
C1rdeficiency SLE-likesyndrome,rheumatoidsyndrome,multiple autoimmunediseases,infections
AR C1r 216950
C1sdeficiency SLE-likesyndrome;multipleautoimmunediseases AR C1s 120580
C4deficiency SLE-likesyndrome,rheumatoidsyndrome, infections
AR C4A 120810
C2deficiency SLE-likesyndrome,vasculitis,atherosclerosis, polymyositis,pyogenicinfections;
glomerulonephritis
AR C2 217000
C3deficiency Lifethreateningpyogenicinfections;SLE-like disease;glomerulonephritis;atypical hemolytic–uremicsyndrome
AR C3 120700
C5deficiency Neisseriainfection,SLE AR C5␣orC5 120900
C6deficiency Neisseriainfection,SLE AR C6 217050
C7deficiency Neisseriainfection,SLE,vasculitis AR C7 217070
C8adeficiency Neisseriainfection,SLE AR C8␣ 120950
C8bdeficiency Neisseriainfection,SLE AR C8 120960
FactorIdeficiency Recurrentpyogenicinfections,glomerulonephritis, SLE;hemolytic–uremicsyndrome;severe
pre-eclampsia
AR CFI 610984
AD,autosomaldominantinheritance;AR,autosomalrecessiveinheritance;SLE,systemiclupuserythematosus.
whichserveastriggersforbreakingimmunetolerance. How-ever,theassociationbetweenSIgADandSLEisnotcompletely understoodyet.
Hyper-IgMsyndrome
Hyper-IgMsyndrome(HIGM)isanon-classicalPID character-izedbyantibodydeficiencywiththeabsenceofIgGandIgA butnormalorincreasedIgMlevels. Differentgenetic muta-tionscancausethisPID;includingmutationofCD40ligand gene(CD40LGgene,X-linkedHIGM),CD40gene, Activation-induced DNA-cytidine deaminase gene (AICDA gene, also known as AID) and uracil DNA glycosylase gene (UNG) (Table 2).19,29 Patients with HIGM usually present during childhood opportunistic infections and autoimmune dis-eases(autoimmunecytopenia,nephritis,inflammatorybowel disease, autoimmune hepatitis, arthritis, hypothyroidism andSLE).Autoimmunemanifestationare morefrequentin patientsthatpresentHIGMduetomutationsinAID,however, autoimmunemanifestationshavealsobeenreportedinother typesofHIGM.29,30 Thereareveryfewcasesreportedonthe coexistenceofSLEandAIDorUNGassociatedHyper-IgM.31
IsolatedIgGsubclassdeficiency
IgG subclassdeficiency isdefined asa serum IgG subclass level thatis morethan two standarddeviations belowthe normalmean forage. IgGsubclassdeficiency canbe asso-ciated with recurrent infections of the upper and lower respiratorytracts.32Pathogensaregenerallylimitedto bacte-riaandrespiratoryviruses.BecauseIgG2isimportantinthe responsetopolysaccharideantigens,IgG2subclass-deficient patientstypicallyhaveinfectionswithHaemophilusinfluenzaor
Streptococcuspneumoniae.33 Inadults,deficiencyofIgG3 sub-classisthemostcommon,whereasinchildrenIgG2isthe mostprevalentIgGsubclassdeficiency.IgGsubclassdeficiency may be seen in conjunction with other primary immune deficiency disorders,such as ataxia-telangiectasia and IgA deficiency.34AnIgGsubclassdeficiencymightoccurasan iso-latedsingleIgGsubclassdeficiencyorasadeficiencyoftwoor moreIgGsubclasses.Theliteraturedescribessporadiccases ofautoimmunemanifestationinpatientswithIgGsubclass deficiency,35–38likeIgG1,39IgG440andcombinedIgG2andIgG4 subclassdeficiency.41Theprevalencemightbehigher, how-ever those casesmight go unnoticed,since IgG subclasses serum level determination isnot includedin routine eval-uation of SLEpatients. Jesus et al. (2011) also showed the coexistence of IgG2 deficiency in 5.5% ofthe juvenile SLE patients studied,representing 21% ofall PID casesintheir series.18
IgMdeficiency
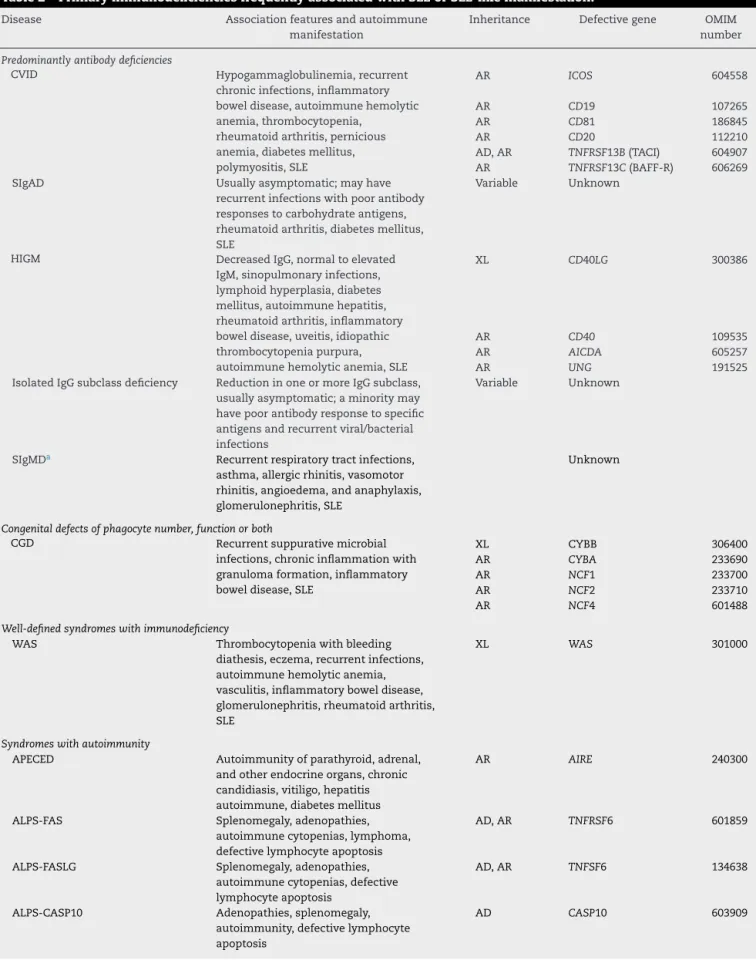
Table2–PrimaryimmunodeficienciesfrequentlyassociatedwithSLEorSLE-likemanifestation.
Disease Associationfeaturesandautoimmune
manifestation
Inheritance Defectivegene OMIM
number
Predominantlyantibodydeficiencies
CVID Hypogammaglobulinemia,recurrent
chronicinfections,inflammatory boweldisease,autoimmunehemolytic anemia,thrombocytopenia,
rheumatoidarthritis,pernicious anemia,diabetesmellitus, polymyositis,SLE
AR ICOS 604558
AR CD19 107265
AR CD81 186845
AR CD20 112210
AD,AR TNFRSF13B(TACI) 604907 AR TNFRSF13C(BAFF-R) 606269
SIgAD Usuallyasymptomatic;mayhave
recurrentinfectionswithpoorantibody responsestocarbohydrateantigens, rheumatoidarthritis,diabetesmellitus, SLE
Variable Unknown
HIGM DecreasedIgG,normaltoelevated
IgM,sinopulmonaryinfections, lymphoidhyperplasia,diabetes mellitus,autoimmunehepatitis, rheumatoidarthritis,inflammatory boweldisease,uveitis,idiopathic thrombocytopeniapurpura, autoimmunehemolyticanemia,SLE
XL CD40LG 300386
AR CD40 109535
AR AICDA 605257
AR UNG 191525
IsolatedIgGsubclassdeficiency ReductioninoneormoreIgGsubclass, usuallyasymptomatic;aminoritymay havepoorantibodyresponsetospecific antigensandrecurrentviral/bacterial infections
Variable Unknown
SIgMDa Recurrentrespiratorytractinfections,
asthma,allergicrhinitis,vasomotor rhinitis,angioedema,andanaphylaxis, glomerulonephritis,SLE
Unknown
Congenitaldefectsofphagocytenumber,functionorboth
CGD Recurrentsuppurativemicrobial
infections,chronicinflammationwith granulomaformation,inflammatory boweldisease,SLE
XL CYBB 306400
AR CYBA 233690
AR NCF1 233700
AR NCF2 233710
AR NCF4 601488
Well-definedsyndromeswithimmunodeficiency
WAS Thrombocytopeniawithbleeding
diathesis,eczema,recurrentinfections, autoimmunehemolyticanemia, vasculitis,inflammatoryboweldisease, glomerulonephritis,rheumatoidarthritis, SLE
XL WAS 301000
Syndromeswithautoimmunity
APECED Autoimmunityofparathyroid,adrenal,
andotherendocrineorgans,chronic candidiasis,vitiligo,hepatitis autoimmune,diabetesmellitus
AR AIRE 240300
ALPS-FAS Splenomegaly,adenopathies,
autoimmunecytopenias,lymphoma, defectivelymphocyteapoptosis
AD,AR TNFRSF6 601859
ALPS-FASLG Splenomegaly,adenopathies,
autoimmunecytopenias,defective lymphocyteapoptosis
AD,AR TNFSF6 134638
ALPS-CASP10 Adenopathies,splenomegaly,
autoimmunity,defectivelymphocyte apoptosis
Table2–(Continued)
Disease Associationfeaturesandautoimmune
manifestation
Inheritance Defectivegene OMIM
number
Caspase8defect Adenopathies,splenomegaly,recurrent bacterial/viralinfections,defective lymphocyteapoptosis,
hypogammaglobulinemia
AD NRAS 164790
Others
ICLa CD4+lymphocytopenia,opportunistic
infections,antiphospholipidsyndrome, psoriasis,Hashimoto’sthyroiditis,Graves disease,ulcerativecolitis,vitiligoandSLE
UNC119
AD,autosomaldominantinheritance;AR,autosomalrecessiveinheritance;XL,X-linkedinheritance;CVID,commonvariable immunodefi-ciency;SIgMD,selectivedeficiencyIgM;SIgAD,selectivedeficiencyIgA;HIGM,hyper-IgMsyndrome;CGD,chronicgranulomatousdisease; WAS,Wiskott–AldrichSyndrome;APECED,autoimmunepolyendocrinopathywithcandidiasisandectodermaldystrophy;ALPS,autoimmune lymphoproliferativesyndrome;ICL,idiopathicCD4+lymphocytopenia;SLE,systemiclupuserythematosus.
a Non-classicPID.
maypresentantinuclearantibodies(ANA).45Fewreportshave focusedtheiranalysisonselectiveIgMdeficiencyhowevera detailedpathogenesisofthisdisorderstillremainstobe care-fullyanalyzed.Astudyonacasereportofa37-year-oldwoman whopresentedselectiveIgMdeficiencywithconcurrentIgG4 deficiency,variousdermalsymptomsandabronchialpolyp, serves as an observation of a not very clear association betweena solitarypolyp and IgMdeficiency, however sug-gestionshavebeenmadeforrepeatedIgMdeficiency-related airway infections as a probable etiological factor for the inflammatorypolyp.46Itisspeculatedthatthe reductionof secretedIgMproductionisrelatedtotheriskofprogression of autoimmune diseases, such as autoimmune glomeru-lonephritisand SLEin humans.Infact, a fewcase reports orseriesshowassociationofthisdisorderwithautoimmune rheumaticdiseases,includingSLE,44,47especiallyinpatients withdiseaseoflongduration.48Interestingly,disease remis-sion did not correlate with elevation ofIgM serum levels, indicatingadeeperdysregulationoftheimmunesystem.
Congenitaldefectsofphagocyte
Phagocytessuchasmonocytes/macrophagesaswellas gra-nulocytes are the cells that engulf and destroy ingested pathogensduringaprocessdenominatedphagocytosis.In cer-tainconditions,eitherthenumberofphagocytesisreduced or their functional capacity is impaired.49 Almost all PID due to phagocyte defects are a consequence of inherited mutations affecting the innate immune system. Most of these PID patients are identified at very young age based on their clinical phenotype of susceptibility to normally nonpathogenicbacteriaorfungi,andinsomecases,the infec-tious agents point tothe disorder.50 Defects ofthese cells includedecreasednumberofneutrophilscausedbydefects ongranulocytedevelopmentorcapabilitytoexitintothe cir-culationleading to neutropenia; or dueto the presenceof autoantibodiesor isoantibodies directedagainstneutrophil membrane antigens. Other defects include abnormalities in granulocyte killing ability, opsonic capability secondary to deficiencies of antibody and complement factors, and chemotaxis.50
Chronicgranulomatousdisease
Chronicgranulomatousdisease(CGD)isaprimary immuno-deficiencyofphagocytes,withX-linkedorautosomalrecessive inheritance. TheX-linked formpresents mutation inCYBB genethatencodestheheavychainofcytochromeb558,or
gp91-phox(56%ofcases),anelectrontransportproteinresponsible fortheoxidativeburstofphagocytes.Thesepatientspresent severe andrecurrentinfections ofskin,respiratory system, gastrointestinal tractand adjacent lymphonodes,pancreas, bonesandcentralnervoussystem.Persistenceof microorgan-ismsinphagolysosomesleadstogranulomaformationthat causesobstructionalongthegastrointestinalorurinarytract. Intheautosomalrecessiveform,genesaffectedincludethe othercomponentsofNADPHoxidasesystem:NCF1(adapter proteinp47-phox,33%ofcases);NCF2(activatorprotein p67-phox, 5% of cases); and NCF4 (p40-phox), 6% of cases.51 PatientswithX-linkedformpresentsevereinfectionsinthe first yearoflife,andpatientswiththe autosomalrecessive formofCGDhavelesssevereclinicalmanifestation,withlate onsetsymptoms.Oralulcersandautoimmunemanifestation (antiphospholipid syndrome,recurrent pericardial effusion, juvenileidiopathicarthritis,IgAnephropathy,cutaneousand systemiclupuserythematosus,andautoimmunepulmonary disease) are frequently seen in patients withCGD.52 Addi-tionally,the mother’sstatus ofcarrier ofthe affected gene isassociatedtohigherfrequencyofdiscoidlupuslesions.11 X-linkedformcanalsopresentMcLeodphenotype(agenetic disorderthatmayaffecttheblood,brain,peripheralnerves, muscleandheart,causedbyavarietyofrecessivelyinherited mutationsintheXKgeneontheXchromosome,responsible forproducingtheKxprotein,asecondarysupportiveprotein fortheKellantigenontheredbloodcellsurface,with compen-satedhemolysis,acanthocysisandprogressivedegenerative neuromusculardisorders).53
Well-definedPIDandPIDsyndromesassociatedwithSLE
haveleadtotheidentificationthatassociationsofPIDwith AIDsaremorefrequentthanpreviouslyappreciated.5,54,55It becameevidentthatdifferenttypesofPIDdisplayconsistent associationswithdistinct autoimmunedisorders(including homozygousdeficienciesofearlycomponentsoftheclassical Complement pathway, selective and partial immunoglobu-lindeficiencies,particularlyisolatedIgAandIgMdeficiencies, andX-linkedandautosomalformsofchronicgranulomatous disease),allowingtheperceptionthatthestudyofthe associ-ationbetweenPIDandAIDsrepresentsauniqueopportunity fornewinsightsandabetterunderstandingofthe pathophy-siologyaswellasthegeneticbasisofautoimmunity.
Wiskott–Aldrichsyndrome
TheWiskott–Aldrichsyndrome(WAS)isaPIDcausedby muta-tionintheWASgene,that encodesaproteinassociatedto theprocess ofcell locomotion,immunological synapse for-mation,apoptosisand phagocytosis.Mutationsinthe WAS gene can lead to severe clinical manifestations (classical WAS),lightmanifestations(X-linkedthrombocytopenia/XLT) and X-linked neutropenia (neutropenia and thrombocyto-peniawithoutmyelodysplasiaorimmunodeficiency).Patients usuallypresentelevatedIgAandIgEserumlevels,normalIgG, andslightlydecreasedIgM.ThecytotoxicactivityofNKcells andCD8Tlymphocytesisimpaired.Infectionsarecommon sincesixmonthsofage,withthedevelopmentofotitismedia, sinusitis,pneumoniaanddiarrhea.Viralinfectionsare com-mon,especiallyforchickenpox,herpessimplexandmolluscum contagiosum. Clinicalpresentationisnormallyvariable,with symptomsappearingsoonafterbirthorinearlylife.Patients with WAS have small platelets, lacking specific granules, reduced numbers oforganelles inthe cytoplasm,defective plateletaggregationandineffectivethrombocytopoiesis.The appearanceofpetechiae,bruising,bleedingandseverecases ofthrombocytopeniahemorrhageofcentralnervoussystem arefrequent.56,57Eczema,recurrentinfections,autoimmune diseases(hemolyticanemia,vasculitis,nephropathy,purpura resemblingHenoch–Schonlein,inflammatoryboweldisease, SLE, and IgA nephropathy) and malignancies (lymphoma, leukemia)arenotraremanifestations.13,58
Autoimmunepolyendocrinopathywithcandidiasis andectodermaldystrophy
Autoimmune polyendocrinopathy candidiasis and ectoder-mal dystrophy (APECED) or autoimmune polyendocrine syndrometypeI(APS1)isaPIDthatharborsautoimmunity withinitsveryessence.Thereisawidevariationintheclinical featuresandcourseofAPECED,evenamongpatientssharing thesamemutationintheautoimmuneregulatorgene(AIRE), involvedinthedisorderandwhoseencodedproteinis respon-sibleforpresentingseveralself-antigensinthymusmedullae. While specific mutations in the AIRE gene have not been associatedwithdiseasephenotype,associationswithspecific HLAhaplotypes have been noted forsome ofthe autoim-mune manifestations of APECED, including alopecia, T1D, andAddison’sdisease.Chronicmucocutaneouscandidiasis, hypoparathyroidism,andadrenocorticalfailurearethe clas-sic triad of findings that characterize this syndrome.59
Otherautoimmuneendocrinopathiescanbepresent, includ-ing insulin-dependent diabetes mellitus, autoimmune thy-roiditis, premature ovarian failure, and hypergonadotropic hypogonadism.Immune-mediatedgastrointestinaldiseases, autoimmunedermatologicconditions,ectodermaldystrophy, keratoconjunctivitis,iridocyclitis,hemolyticanemia,oraland esophagealcancers,chronichepatitis,nephritis,cholelithiasis andSLEhavealsobeenseenassociatedtoAPECED.60,61
Autoimmunelymphoproliferativesyndrome
Autoimmune lymphoproliferative syndrome (ALPS) is an autosomal dominant disorder caused by abnormalities in Fas-mediated lymphocyte apoptosis, with clinical fea-tures of splenomegaly and lymphadenopathy, and various autoimmune manifestations. ALPS causedbyheterozygous mutations in the Fas gene (TNFRSF6; ALPS Type Ia) make up the majority ofidentified cases. ALPS caused by muta-tionsinotherfactorsinvolvedintheFasapoptosispathway havebeenidentified,includingFasL(TNFSF6;ALPSTypeIb), Caspase 8(NRAS)and Caspase 10 (CASP10) (the lattertwo, ALPS Type II). There is also a subgroup of patients with ALPS phenotype, abnormalFas-mediated apoptosis,but no identified mutationinthe Fas pathway(ALPS Type III).62,63 ImmunologicalabnormalitiescharacteristicofALPSinclude the presenceofincreasednumber ofcirculating CD4−CD8−
␣/+ lymphocytes (double negative), as well as T- and B-celllymphocytosisandpolyclonalhypergammaglobulinemia. Autoimmune hemolytic anemia and immune thrombocy-topenia are the most common autoimmune features seen in ALPS. Autoimmune neutropenia and the presence of anticardiolipin antibodies are also often present, whereas autoimmune hepatitis, uveitis, and glomerulonephritis are muchlesscommonmanifestationsinthesepatients.63The literaturedescribesacaseofSLE-likesyndromeina 59-year-oldwomanwitharthritis,lowfever,intermittenthypotension, confusion,macularskinrashwithtelangiectasiaand perivas-cular lymphocyte infiltration, cytopenia without abnormal cells,hepatosplenomegaly, pericardialand pleural effusion, cervical lymph node enlargements and diffuselargeB cell lymphoma. This patient was described with autoimmune lymphoproliferativesyndrome-likesyndrome.15
IdiopathicCD4+lymphocytopenia
Idiopathic CD4+ lymphocytopenia (ICL) is a non-classical
PIDcharacterizedbyaTCD4+ lymphocytecellcountbelow
300/mm3 or 20% of total T lymphocyte cell count in the
absence ofidentifiedcause,including human immunodefi-ciency virus(HIV) orhumanlymphocytotropicvirus(HTLV) infections, and absence of causative drug.64 Recently, a mutation in patients with ICL was described,65 but fur-ther studies are needed fordefinitive conclusion sincethe etiology still remains poorly understood and inadequately defined.MechanismsimplicatedinCD4+ lymphocyte
anoncogenicpotential(humanpapillomavirus/HPV,Kaposi’s sarcomabyHHV8+).66,67 Autoimmunediseases observed in a series of 39 cases of ICL include SLE, antiphospholipid syndrome,psoriasis,Hashimoto’sthyroiditis,Gravesdisease, ulcerativecolitisandvitiligo.68,69
ClinicalcharacteristicsofpatientswithSLEand PIDmanifestationsfollowedbytheoutpatient clinicoftheRheumatologyDivision
atUniversidadeFederaldeSãoPaulo
Between2009and2011,ourgroupfollowed315consecutive adultSLEpatientsattheRheumatologyDivisionoutpatient clinicofthe UniversityHospitalofUniversidadeFederalde SãoPaulo. Thepurposeofthe study wasto systematically trackacomprehensivearrayofPIDinalargecohort.Oncethe diseaseactivitycouldinfluencetheresults,allpatientswere followeduntilachievingdiseasequiescence.Fifteenpatients remainedwithactivediseasethroughoutthefollow-upand were,therefore,excludedfromtheanalysis.Patientsfollowed were predominantly females (16 males and 284 females), with 39.58±12.54 mean years-old (age ranging from 18 to 61years),meandiseasedurationof10.74±8.15years(disease durationfrom 1to53 years)andmean ageatSLEonset of 28.79±10.89years-old (SLEonset from 3to69 years).Total frequencyofinfectionsinSLEpatientswas28(9.33%).Those patients were classified using the warningsignals for pri-maryimmunodeficiencyrecentlyrevised.70Unfortunatelythe cross-sectionaldesignofourstudycouldnotallowthecalculi ofmortalityrate.Ninepatientshadrecurrentairway infec-tions,whereas15presentedrecurrenturinarytractinfections andthree,skinfurunculosis.Twopatientspresentedrecurrent oral/genitalHerpessimplexand twoothershadHerpeszoster infection.Additionally,twopatientsmanifested mycobacte-rialinfection:onehadpulmonarytuberculosisandtheother hanseniasis(Table3).Inthepresentseries,otherautoimmune diseaseswereobservedin47individuals(15.66%)[including rheumaticautoimmunediseases(n=32)andnon-rheumatic autoimmunediseases(n=20)],someofwhichpresentingmore thanoneautoimmunecondition.Eighty-fourpatients(28%) wereidentifiedwithimmunitydefectscompatiblewith clas-sical PID (Table 3), and in four patients (1.3%) more than oneassociatedPIDwereidentified(SIgAD+IgG2;SIgAD+IgG4; IgMD+IgG2in2patients).Differentlyfromourresults,the lit-eraturedescribesonecaseofSIgMDaccompaniedwithIgG4 deficiency(Ideuraetal.46).Interestingly,onepatientpresented arespiratoryburstprofileimpaired enoughtobeclassified asaCGD genecarrierbut no patientpresentedthe profile compatible with full-blown disease. Our clinical and labo-ratory findings have demonstrated that the PID observed inSLEpatientsare considered mildinterms ofseverityof infectionsandmortality.WespeculatethatthosePIDare com-patiblewithapparentlynormallife,butthattheconsequent long-standingantigenicburdenmaybeariskfactorforthe developmentofAIDs,representedinthiscohortbySLE. Gen-erally,severe formsof PIDare diagnosed atearly stage of life,whilenon-severeormildformsofPIDmanifestationsare mostlyasymptomatic.20Wefoundthat28%ofourcohortof adultSLEpatientswasconstitutedbymildPIDwhichallowed alongersurvivalrate, passingunnoticedduringchildhood.
Table3–Autoimmunediseases,primary immunodeficienciesandinfectionsfoundin300 BrazilianSLEpatients.
Autoimmunerheumaticdiseasen=32(10.6%)
Antiphospholipidsyndrome n=16(5.3%)
Sjögrensyndrome n=7(2.3%)
Systemicsclerosis n=4(1.3%)
Rheumatoidarthritis n=4(1.3%)
Polymyositis n=2(0.6%)
Psoriaticarthritis n=1(0.3%)
Non-rheumaticautoimmunediseasen=20(6.6%)
Hypothyroidism n=15(5%)
Psoriasis n=3(1%)
Vitiligo n=3(1%)
Primarybiliarycirrhosis n=1(0.3%)
IgAnephropathy n=1(0.3%)
Primaryimmunodeficiencydisease(Classicalandnon-Classical) n=84(28%)
SIgMDa n=24(8%)
SIgAD n=3(1%)
DefIgG n=1(0.3%)
DefIgG1 n=5(1.6%)
DefIgG2 n=40(13.3%)
DefIgG3 n=24(8%)
DefIgG4 n=11(3.6%)
CGDgenecarrier n=1(0.3%)
Infectionsn=28(9.33%)
Airwayinfection n=9(3%)
Urinarytractinfection n=15(5%)
Furunculosis n=3(1%)
Herpessimplex n=2(0.6%)
Herpeszoster n=2(0.6%)
Tuberculosis n=1(0.3%)
Hanseniasis n=1(0.3%)
SIgAD,selectiveIgAdeficiency;SIgMD,selectiveIgMdeficiency.
a Non-classicPID.
Thiscouldpossiblyexplaintheabsenceofillnessessuchas CVID,CGDandHIGM.Surprisingly,inourcohortthepresence ofIgMD,anon-classicalformofPID,wasveryfrequent.Wealso observedinourcohortalargenumberofSLEpatientswithIgG subclassdeficiency,whileliteraturereportsonlysomecases ofisolateddeficiencyofIgG2andIgG4.18,41,71,72Inourstudy, allpatientswithIgG4deficiencyand75%ofthosewithIgG3 deficiencyhadlupusnephropathy,whichisabovethe∼50% frequencyinthewholecohort.Inaddition,patientswithIgMD presentedlowerfrequencyoforalulcers.ApartfromIgG4and IgG3deficientpatients,theremainingpatientsdidnotpresent amuchseverephenotyperegardingthepresenceofinfections andlupusmanifestations.
unbalancedIgGsubclassessynthesis,whichmaybe consid-eredas afactor forthe developmentofSLE. These results suggestthatmildimmunologicdefectsmightbecompatible withpatientsurvival,butattheexpenseofsomechronic over-loadandfutureconsequencestotheimmunesystem,which couldleadtothedevelopmentofimmunedisorders character-isticofSLEintheadulthood.Thestudyfindingsgivegroundto furtherinvestigationsthatcoulddeeplyexplorethe participa-tionofPIDinthepathogenesisofSLEandotherautoimmune rheumaticandnon-rheumaticdisease.
Conclusion
PIDareagroupofmonogenic diseasesinwhichmutations ofcertaingenescanleadtoincreasedsusceptibilityto infec-tionsbutmayalsoresultinlossofcentraland/orperipheral tolerance.Therefore,AIDsarecommonamongpatientswith adiverse array of PID.Immunoglobulin deficiency forms a peculiargroupofPID,inwhichtheinheritanceappearstobe polygenicand thereisawide severityspectrum,withmild formsthat usuallyremain unnoticed.Ourfindingsinadult patientswithSLEsuggestthatAIDscanpresentahigher fre-quencyoflesssevereformsofPIDwithoutsevereinfections. ThepresenceofsomeformsofPIDwasassociatedwith cer-tainphenotypic peculiaritiesinSLEpatients.Theliterature andourfindingsshowthatPIDandAIDsfrequentlycoexist andpatientswithautoimmunediseasesshouldbecarefully monitoredforthepresenceofPIDandviceversa.
Funding
Fundac¸ão de Amparo a Pesquisa do Estado de São Paulo (FAPESP);ConselhoNacionalde Desenvolvimento Científico eTecnológico(CNPq); Coordenac¸ão deAperfeic¸oamento de PessoaldeNívelSuperior(CAPES).
Conflicts
of
interest
Theauthorsdeclarenoconflictsofinterest.
r
e
f
e
r
e
n
c
e
s
1. GuptaS,LouisAG.Toleranceandautoimmunityinprimary immunodeficiencydisease:acomprehensivereview.ClinRev AllergyImmunol.2013;45:162–9.
2. MoroniL,BianchiI,LleoA.Geoepidemiology,gender,and autoimmunedisease.AutoimmunRev.2012;11:A386–92. 3. TorgersonTR.Immunodeficiencydiseaseswithrheumatic
manifestations.PediatrClinNAm.2012;59:493–507. 4. RahmanA,IsenbergDA.Systemiclupuserythematosus.N
EnglJMed.2008;358:929–39.
5. ArasonGJ,JorgensenGH,LudvikssonBR.Primary
immunodeficiencyandautoimmunity:lessonsfromhuman diseases.ScandJImmunol.2010;71:317–28.
6. DooleyMA,HoganSL.Environmentalepidemiologyandrisk factorsforautoimmunedisease.CurrOpinRheumatol. 2003;15:99–103.
7.MandersonAP,BottoM,WalportMJ.Theroleofcomplement inthedevelopmentofsystemiclupuserythematosus.Annu RevImmunol.2004;22:431–56.
8.CassidyJT,KitsonRK,SelbyCL.SelectiveIgAdeficiencyin childrenandadultswithsystemiclupuserythematosus. Lupus.2007;16:647–50.
9.Carneiro-SampaioM,LiphausBL,JesusAA,SilvaCA,Oliveira JB,KissMH.Understandingsystemiclupuserythematosus physiopathologyinthelightofprimaryimmunodeficiencies. JClinImmunol.2008;28Suppl.1:S34–41.
10.RosenzweigSD.Inflammatorymanifestationsinchronic granulomatousdisease(CGD).JClinImmunol.2008;28Suppl. 1:S67–72.
11.WinkelsteinJA,MarinoMC,JohnstonRBJr,BoyleJ,CurnutteJ, GallinJI,etal.Chronicgranulomatousdisease.Reportona nationalregistryof368patients.Medicine(Baltimore). 2000;79:155–69.
12.CaleCM,MortonL,GoldblattD.Cutaneousandother lupus-likesymptomsincarriersofX-linkedchronic
granulomatousdisease:incidenceandautoimmuneserology. ClinExpImmunol.2007;148:79–84.
13.MonteferranteG,GianiM,vandenHeuvelM.Systemiclupus erythematosusandWiskott–AldrichsyndromeinanItalian patient.Lupus.2009;18:273–7.
14.ChebbiW,AlayaW,ZantourB,BerricheO,KamounM,Sfar MH.Systemiclupuserythematosuswithautoimmune polyendocrinopathytypeII.PresseMed.2011;40: 772–4.
15.HongYH,LeeCK.Autoimmunelymphoproliferative syndrome-likesyndromepresentedaslupus-likesyndrome withmycobacterialjointinfectionevolvedintothe lymphoma.RheumatolInt.2009;29:569–73.
16.CoutantG,AlgayresJP,BiliH,DalyJP.CD4lymphocytopenia. Gougerot–Sjogrenandsystemiclupuserythematosus.Ann MedIntern(Paris).1997;148:503–4.
17.ListonA,EndersA,SiggsOM.Unravellingtheassociationof partialT-cellimmunodeficiencyandimmunedysregulation. NatRevImmunol.2008;8:545–58.
18.JesusAA,LiphausBL,SilvaCA,BandoSY,AndradeLE, CoutinhoA,etal.Complementandantibodyprimary immunodeficiencyinjuvenilesystemiclupuserythematosus patients.Lupus.2011;20:1275–84.
19.Al-HerzW,BousfihaA,CasanovaJL,ChapelH,ConleyME, Cunningham-RundlesC,etal.Primaryimmunodeficiency diseases:anupdateontheclassificationfromthe internationalunionofimmunologicalsocietiesexpert committeeforprimaryimmunodeficiency.FrontImmunol. 2011;2:54.
20.CasanovaJL,FieschiC,BustamanteJ,ReichenbachJ,RemusN, vonBernuthH,etal.Fromidiopathicinfectiousdiseasesto novelprimaryimmunodeficiencies.JAllergyClinImmunol. 2005;116:426–30.
21.MizunoM.Areviewofcurrentknowledgeofthecomplement systemandthetherapeuticopportunitiesininflammatory arthritis.CurrMedChem.2006;13:1707–17.
22.GlesseN,MonticieloOA,MatteviVS,BrenolJC,XavierRM,da SilvaGK,etal.Associationofmannose-bindinglectin2gene polymorphicvariantswithsusceptibilityandclinical progressioninsystemiclupuserythematosus.ClinExp Rheumatol.2011;29:983–90.
23.PandaAK,ParidaJR,TripathyR,PattanaikSS,RavindranB, DasBK.Mannosebindinglectin:abiomarkerofsystemic lupuserythematosusdiseaseactivity.ArthritisResTher. 2012;14:R218.
25.Cunningham-RundlesC.Themanyfacesofcommonvariable immunodeficiency.HematolAmSocHematolEducProgr. 2012;2012:301–5.
26.Fernandez-CastroM,Mellor-PitaS,CitoresMJ,MunozP, Tutor-UretaP,SilvaL,etal.Commonvariable
immunodeficiencyinsystemiclupuserythematosus.Semin ArthritisRheum.2007;36:238–45.
27.AgarwalS,Cunningham-RundlesC.Autoimmunityin commonvariableimmunodeficiency.CurrAllergyAsthma Rep.2009;9:347–52.
28.WangN,ShenN,VyseTJ,AnandV,GunnarsonI,SturfeltG, etal.SelectiveIgAdeficiencyinautoimmunediseases.Mol Med.2011;17:1383–96.
29.UygungilB,BonillaF,LedermanH.Evaluationofapatient withhyper-IgMsyndrome.JAllergyClinImmunol. 2012;129:1692e4–3e4.
30.BussoneG,MouthonL.Autoimmunemanifestationsin primaryimmunedeficiencies.AutoimmunRev.2009;8: 332–6.
31.MelegariA,MasciaMT,SandriG,CarbonieriA.
Immunodeficiencyandautoimmunephenomenainfemale
hyper-IgMsyndrome.AnnNYAcadSci.2007;1109:106–8. 32.AgarwalS,Cunningham-RundlesC.Assessmentandclinical
interpretationofreducedIgGvalues.AnnAllergyAsthma Immunol.2007;99:281–3.
33.MaguireGA,KumararatneDS,JoyceHJ.Arethereanyclinical indicationsformeasuringIgGsubclasses?AnnClinBiochem. 2002;39:374–7.
34.AghamohammadiA,CheraghiT,GharagozlouM,Movahedi M,RezaeiN,YeganehM,etal.IgAdeficiency:correlation betweenclinicalandimmunologicalphenotypes.JClin Immunol.2009;29:130–6.
35.DuzgunN,PeksariY,SonelB,YucesanC,ErekulS,DumanM. Localizationofextrapulmonarytuberculosisinthesynovial membrane,skin,andmeningesinapatientwithsystemic lupuserythematosusandIgGdeficiency.RheumatolInt. 2002;22:41–4.
36.OxeliusVA.ImmunoglobulinG(IgG)subclassesandhuman disease.AmJMed.1984;76:7–18.
37.DuzgunN,DumanM,SonelB,PeksariY,ErdemC,TokgozG. Lupusvulgarisinapatientwithsystemiclupus
erythematosusandpersistentIgGdeficiency.RheumatolInt. 1997;16:213–6.
38.VisitsunthornN,HengcrawitW,JirapongsananurukO, LuangwedchakamV.ImmunoglobulinG(IgG)subclass deficiencyinThaichildren.AsianPacificJAllergyImmunol. 2011;29:332–7.
39.LacombeC,AucouturierP,Preud’hommeJL.SelectiveIgG1 deficiency.ClinImmunolImmunopathol.1997;84:194–201. 40.KimJH,ParkHJ,ChoiGS,KimJE,YeYM,NahmDH,etal.
ImmunoglobulinGsubclassdeficiencyisthemajor phenotypeofprimaryimmunodeficiencyinaKoreanadult cohort.JKoreanMedSci.2010;25:824–8.
41.TamuraA,AgematsuK,UrasawaR,NaganumaK,Komiyama A.Cardiactamponadeduetosystemiclupuserythematosus ina7-year-oldboywithselectiveIgGsubclassdeficiency.EurJ Pediatr.1998;157:475–8.
42.Al-HerzW,McGeadySJ,GrippKW.22q11.2deletionsyndrome andselectiveIgMdeficiency:anassociationofacommon chromosomalabnormalitywitharareimmunodeficiency.Am JMedGenetA.2004;127A:99–100.
43.KungSJ,GrippKW,StephanMJ,FairchokMP,McGeadySJ. SelectiveIgMdeficiencyand22q11.2deletionsyndrome.Ann AllergyAsthmaImmunol.2007;99:87–92.
44.GoldsteinMF,GoldsteinAL,DunskyEH,DvorinDJ,Belecanech GA,ShamirK.SelectiveIgMimmunodeficiency:retrospective analysisof36adultpatientswithreviewoftheliterature.Ann AllergyAsthmaImmunol.2006;97:717–30.
45.AntarM,LamarcheJ,PegueroA,ReissA,ColeS.Acaseof selectiveimmunoglobulinMdeficiencyandautoimmune glomerulonephritis.ClinExpNephrol.2008;12:300–4. 46.IdeuraG,AgematsuK,KomatsuY,HatayamaO,YasuoM,
TsushimaK,etal.SelectiveIgMdeficiencyaccompaniedwith IgG4deficiency,dermalcomplications,andabronchialpolyp. AllergolInt.2008;57:99–105.
47.GoldsteinMF,GoldsteinAL,DunskyEH,DvorinDJ,Belecanech GA,ShamirK.PediatricselectiveIgMimmunodeficiency.Clin DevImmunol.2008;2008:624850.
48.SaikiO,SaekiY,TanakaT,DoiS,HaraH,NegoroS,etal. DevelopmentofselectiveIgMdeficiencyinsystemiclupus erythematosuspatientswithdiseaseoflongduration. ArthritisRheum.1987;30:1289–92.
49.NotarangeloLD,FischerA,GehaRS,CasanovaJL,ChapelH, ConleyME,etal.Primaryimmunodeficiencies:2009update.J AllergyClinImmunol.2009;124:1161–78.
50.Lekstrom-HimesJA,GallinJI.Immunodeficiencydiseases causedbydefectsinphagocytes.NEnglJMed.
2000;343:1703–14.
51.StasiaMJ,LiXJ.Geneticsandimmunopathologyofchronic granulomatousdisease.SeminImmunopathol.
2008;30:209–35.
52.DeRavinSS,NaumannN,CowenEW,FriendJ,HilligossD, MarquesenM,etal.Chronicgranulomatousdiseaseasarisk factorforautoimmunedisease.JAllergyClinImmunol. 2008;122:1097–103.
53.WatkinsCE,LitchfieldJ,SongE,JaishankarGB,MisraN,Holla N,etal.Chronicgranulomatousdisease,theMcLeod phenotype,andthecontiguousgenedeletionsyndrome–a review.ClinMolAllergy.2011;9:13.
54.Carneiro-SampaioM,CoutinhoA.Toleranceand autoimmunity:lessonsatthebedsideofprimary immunodeficiencies.AdvImmunol.2007;95:51–82. 55.WesterbergLS,KleinC,SnapperSB.BreakdownofTcell
toleranceandautoimmunityinprimaryimmunodeficiency– lessonslearnedfrommonogenicdisordersinmiceandmen. CurrOpinImmunol.2008;20:646–54.
56.PuckJM,CandottiF.LessonsfromtheWiskott–Aldrich syndrome.NEnglJMed.2006;355:1759–61.
57.OchsHD,FilipovichAH,VeysP,CowanMJ,KapoorN. Wiskott–Aldrichsyndrome:diagnosis,clinicalandlaboratory manifestations,andtreatment.BiolBloodMarrow
Transplant.2009;151Suppl.:84–90.
58.CatucciM,CastielloMC,PalaF,BosticardoM,VillaA. AutoimmunityinWiskott–Aldrichsyndrome:anunsolved enigma.FrontImmunol.2012;3:209.
59.HalonenM,EskelinP,MyhreAG,PerheentupaJ,HusebyeES, KampeO,etal.AIREmutationsandhumanleukocyteantigen genotypesasdeterminantsoftheautoimmune
polyendocrinopathy-candidiasis-ectodermaldystrophy phenotype.JClinEndocrinolMetab.2002;87:2568–74. 60.PerheentupaJ.Autoimmune
polyendocrinopathy-candidiasis-ectodermaldystrophy.JClin EndocrinolMetab.2006;91:2843–50.
61.SmithCJ,OscarsonM,RonnblomL,AlimohammadiM, PerheentupaJ,HusebyeES,etal.TSGA10–atargetfor autoantibodiesinautoimmunepolyendocrinesyndrometype 1andsystemiclupuserythematosus.ScandJImmunol. 2011;73:147–53.
62.SnellerMC,DaleJK,StrausSE.Autoimmune
lymphoproliferativesyndrome.CurrOpinRheumatol. 2003;15:417–21.
63.TeacheyDT.Newadvancesinthediagnosisandtreatmentof autoimmunelymphoproliferativesyndrome.CurrOpin Pediatr.2012;24:1–8.
aboutidiopathicCD4(+)lymphocytopenia?RevMedIntern. 2012;33:628–34.
65.GorskaMM,AlamR.Consequencesofamutationinthe UNC119geneforTcellfunctioninidiopathicCD4 lymphopenia.CurrAllergyAsthmaRep.2012;12:396–401. 66.RichettaA,AmorusoGF,AscoliV,NataleME,CarboniV,
CarlomagnoV,etal.PEL,Kaposi’ssarcomaHHV8+and idiopathicT-lymphocitopeniaCD4+.ClinTer.2007;158:151–5. 67.AlisjahbanaB,DinataR,SutedjaE,SuryahudayaI,Soedjana
H,HidajatNN,etal.Disfiguringgeneralizedverrucosisinan IndonesianmanwithidiopathicCD4lymphopenia.Arch Dermatol.2010;146:69–73.
68.ZoniosD,SheikhV,SeretiI.IdiopathicCD4lymphocytopenia: acaseofmissing,wanderingorineffectiveTcells.Arthritis ResTher.2012;14:222.
69.ZoniosDI,FalloonJ,BennettJE,ShawPA,ChaittD,Baseler MW,etal.IdiopathicCD4+lymphocytopenia:naturalhistory andprognosticfactors.Blood.2008;112:287–94.
70.Costa-CarvalhoBT,GrumachAS,FrancoJL,Espinosa-Rosales FJ,LeivaLE,KingA,etal.Attendingtowarningsignsof primaryimmunodeficiencydiseasesacrosstherangeof clinicalpractice.JClinImmunol.2014;34:10–22. 71.HansonLA,SoderstromR,AvanziniA,BengtssonU,
BjorkanderJ,SoderstromT.Immunoglobulinsubclass deficiency.PediatrInfectDisJ.1988;75Suppl.: S17–21.