The conjugate addition of a stabilized carbanion nucleophile to an unsaturated carbonyl compound E is called a Michael addition (or Michael reaction), and is considered one of the fundamental reactions of C-C bond formation in organic chemistry.3 Michael addition is a step elegantly on the way to synthesizing more complex molecular frameworks. In modern applications, N-, O-, S-, and P nucleophiles have also attracted considerable attention, especially in asymmetric synthesis.4 The acceptor can be virtually any double or triple bond in the E-bond with an electron-withdrawing group. resonance stabilizers.
The Advent of Asymmetric Organocatalysis
Bifunctional catalysts12 can simultaneously activate both nucleophiles and electrophiles, resulting in a highly organized assembly of the transition state, especially if there are covalent interactions with the catalyst and the substrate, which usually increases the reaction rate and stereoselectivity.9c In this thesis , experimental results related to both categories will be presented, with Chapter 2 focusing on the use of bifunctional H-bond organocatalysis, and Chapter 3 focusing on enamine catalysis, respectively.
Hydrogen Bonding in Catalysis
- Non-Covalent Organocatalysis
- Hydrogen Bonds
- Urea and Thiourea Catalysts
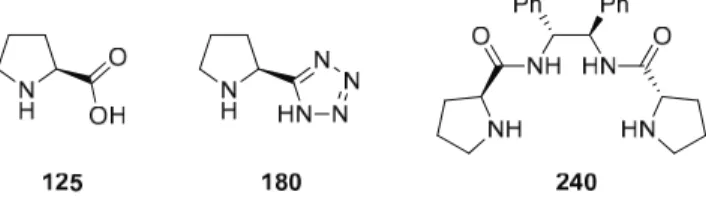
- Cinchona Alkaloid Derived Catalysts
- Thiourea Type Organocatalysts in Target-Oriented Synthesis
The H-bond donor and acceptor can be parts of the same molecule (intramolecular H-bonding) or belong to separate molecules (intermolecular H-bonding). The hydrocyanation of benzaldehyde 36 catalyzed by quinine 37 (or quinidine) afforded a chiral product with enantiomeric excess below 10% (Scheme 8).7 The next landmark was achieved in 1960 when Pracejus published the asymmetric addition of alcohols to phenyl alcohols. ketene 39 catalyzed by O-acetylquinine 40,28. The reaction proceeded in good yields and, consistent with the date, in very good enantioselectivities, up to 74% ee (Scheme 9).

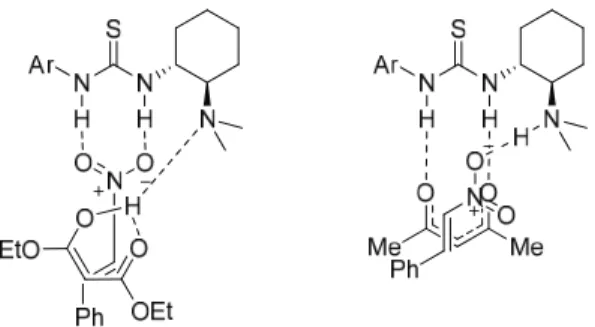
Addition of Meldrum’s Acid to Nitroalkenes
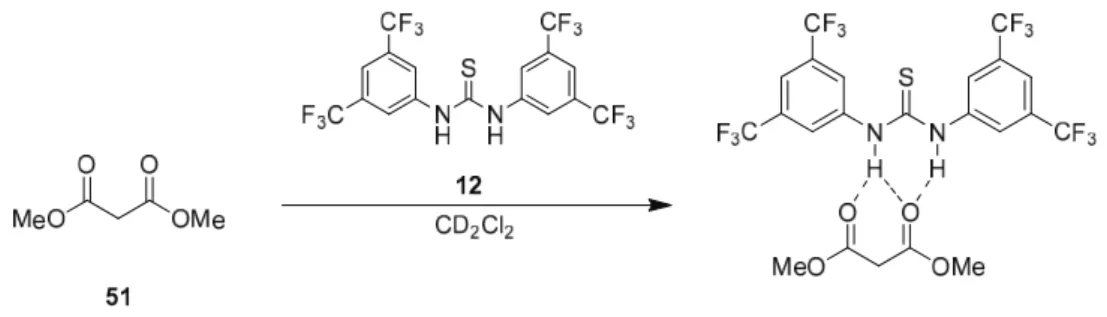
Meldrum's acid is thus capable of forming defined ion pairs with virtually any readily available base. The use of Meldrum's acid as a nucleophile in asymmetric synthesis has been rather limited, and there are several examples of e.g.
Results and Discussion
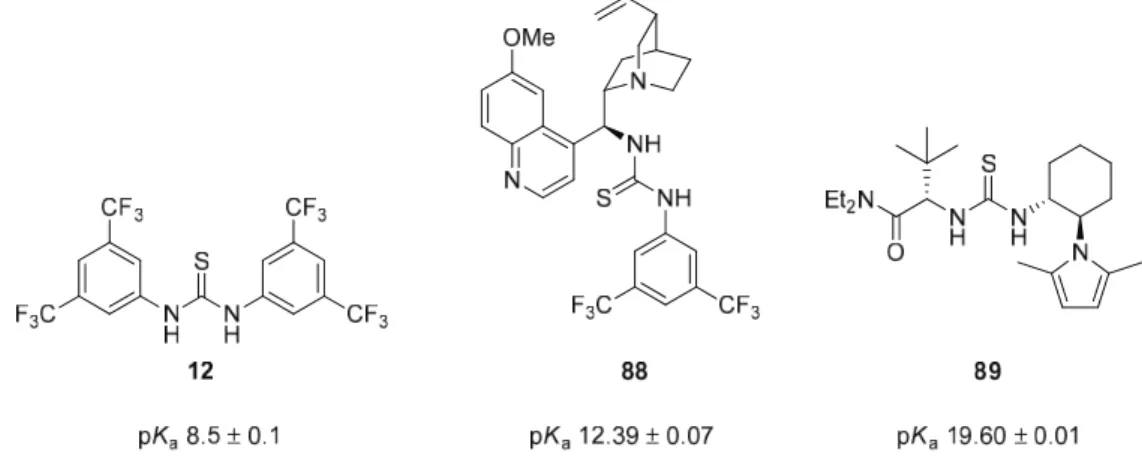
The opposite catalyst 97 was achieved by inverting the isocyanate and amine frameworks.49 First, 90 was treated with CS2 in the presence of DCC to give isothiocyanate 101. Deprotonation of dimethyl malonate in the presence of Schreiner thiourea 12 is very fast.
Conclusions
Four different total syntheses of (–)-henoxazole A have been reported to date and are briefly reviewed in Section 3.3.1.72. An overview of continuous flow technologies, advantages and disadvantages is given below in Section 3.1.

Continuous Flow Technologies
Benefits and Disadvantages of Continuous Flow Conditions
The volume-to-area ratio of continuous flow reactors is typically very high, on the order of m3/m2, which makes heat transfer to and from the reaction system very efficient, depending on the thermal conductivity of the reactor material. When continuous flow conditions are applied to multistep synthesis, monitoring the reaction and coordinating further steps can become problematic (the so-called "third flow problem").
General Strategies for Continuous Flow Synthesis
In the case of asymmetric catalysis, steric effects caused by the structure of the solid support can also deteriorate the selectivity of the catalyst. Most of these issues depend on the synthesis of the solid support (degree of cross-linking in a polymer, polymer beads vs. monolithic column, sol-gel synthesis of SiO2 with controlled pore size, etc.) and the catalyst immobilization method (co- -polymerization, grafting, etc.). The time taken by the synthesis was 8 hours, which is significantly faster than performing the same synthesis by traditional methods, although the rate of flow synthesis is quite small.
The synthesis of oxomaritidine also illustrates one of the major advantages of continuous flow systems.
Covalent Organocatalysis
Enamine Catalysis
The formation of an enamine raises the HOMO energy of the enolate C=C bond and thus increases the -carbon nucleophilicity of a ketone or an aldehyde compared to the bare enol form. For catalysts without a directing H-bond donor or Brønsted acid, such as the Hayashi–Jørgensen type diarylprolinol silyl ethers89, the bulky substituent blocks the other side of the enamine, directing the attack to occur from the unhindered side (Figure 16, II) .90.
Asymmetric Organocatalytic Aldol Reactions
One of the major challenges in organocatalytic aldol reactions is the regulation of selectivity in the intermolecular cross-aldol reaction between two reactive aliphatic aldehyde species. The MacMillan group used both (S)- and (R)-proline in their total synthesis of Callipeltoside C (Scheme 41).104 The first step in the tetrahydropyran fragment synthesis was the aldol reaction between propionaldehyde and Roche aldehyde 152. TIPS-protected -hydroxyacetaldehyde 154 was dimerized using (R)-proline in good yield and 99% ee in the synthesis of the mannose fragment.
To correct this, enantiomeric mannose was synthesized using ( S )-proline in the first step with identical yield and enantioselectivity.
Asymmetric Organocatalysis in Continuous Flow Conditions
At the time of our studies there was only one published report on asymmetric organocatalytic aldol reactions under continuous flow conditions by the Seeberger group.116 They showed the proof of principle of performing an organocatalytic aldol reaction under flow conditions. The current state of asymmetric organocatalytic chemistry in continuous flow is advancing rapidly, and several research groups are investigating the use of polymer or solid-supported reagents. The Pericàs group has recently made impressive advances in aldol and Mannich reactions under continuous flow conditions with polymer-supported prolines.117 Both propionaldehyde and isovaleraldehyde can be added to the glyoxalate imine 182 with high selectivity by pumping the stock solutions of the starting material through a column packed with triazole. bound 4-trans-hydroxy proline 183 on a Merrifield resin (Scheme 46).117a.
In a continuous flow setting, a mixture of 4-nitrobenzaldehyde 128 and cyclohexanone 185 in DMF:H2O (60:40) was pumped through a column packed with 600 mg of swollen resin containing 186 .
Total Synthesis of (–)-Hennoxazole A
- Comparison of Previous Syntheses
- Previous studies by the Ley group
- First Approach to the Synthesis of the Western Fragment
- Second Approach to the Synthesis of the Western Fragment
- Optimization of the Bisoxazole Synthesis
- Catalyst Screening
- Organocatalytic Aldol Step under Continuous Flow Conditions
- Methylation studies
Previous studies in the group had not yielded promising results for this step either. We decided to use the bisoxazole aldehyde 243 as an aldol acceptor partner in the addition of acetone, as this would better match the good results in proline derivative catalyzed aldol reactions. The crude products were also considered pure enough by 1H-NMR analysis for use in the subsequent steps without further purification.
Downfield shift of 1H NMR signal in the closer oxazole unit was observed, along with an upshift of the terminal methyl group, providing preliminary evidence of the desired R configuration following the procedure by Mosher.
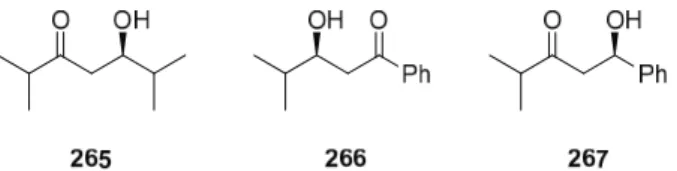
Diastereoselectivity of E -Hydroxy Ketone Reduction with Solid Supported
We also tried to carry out the reaction in a mixture of AcOH and MeOH, but did not observe any reaction. By heating the reaction we were able to significantly increase the reaction rate, without much effect on the diastereoselectivity. When the reaction was carried out in i-PrOH, we observed an anti-enriched product although the reaction was very slow and not as pure as in MeOH (entry 1).
Heating the reaction in i-PrOH gave a faster conversion (not shown in the table), but no product was isolated after filtration, which may be due to the instability of the resin at higher temperatures.
Conclusions
Common aspects of aromatic nitration
Nitration of Vanillin
- Preliminary studies
- Pulse experiments
- Identification of impurities
- Continuous experiments in 5 mL reactor
- Continuous experiments in 10 mL reactor
- Isolation studies and comparison between batch and flow modes
We observed that the conversion of 271 was incomplete, most likely due to co-crystallization with the product during the course of the reaction. In most of the experiments in Table 10, 272 could be observed to precipitate during the course of the reaction. In several cases, the reaction only took place in the collection flask when the entire mixture was collected (series 1-3).
If the concentration was high enough, the reaction in the beaker went to completion and precipitation of the product was observed.

Reaction profile and formation of impurities
However, a survey of the literature reveals that the dealkylation of phenolic ethers under nitration conditions has been previously reported. 136,139 Studies by Ingold established that the concomitant dealkylation is closely related to the reactive nitration species, either nitrosonium or nitronium ion, first. being much more active in the dealkylation process. They ruled out a common acid-promoted demethylation process based on a model experiment in which no significant amounts of dealkylation occurred in the presence of an equimolar amount of H2SO4. Decarbonylative and decarboxylative nitration of electron-rich substrates has also been studied using dilute nitric acid.142 Bentley reported the formation of 2,4-dinitroguaiacol 276 from 5-nitrovanillin 272 when subjected to dilute nitration conditions,137c but in the experiment In our testing we had to use stringent conditions (several equivalents of HNO3, high temperature) before we saw an increase in the amount of 276.
Thus, based on the actual calculated surface area, the increase of 276 in the reaction mixture is quite modest, although obvious.
Nitration of other substrates
We have conducted a brief investigation into the applicability of our relatively mild nitriding process on other substrates. With salicylaldehyde, under similar conditions, we achieved a maximum yield of only 35% with the expected mixture of regioisomers. Anisaldehyde, 4-methoxyacetophenone and 4-methylacetophenone did not react under similar conditions and would clearly require more forcing conditions, e.g.
Although this is possible and has precedent, as shown in Scheme 77, we decided not to explore the topic further as this would stray from our original goal of relatively mild nitration under continuous flow conditions.
Conclusions
The continuous current nitration with a stoichiometric amount of dilute nitric acid is especially applicable to highly reactive substrates, as shown by our short substrate studies.
General Experimental Considerations
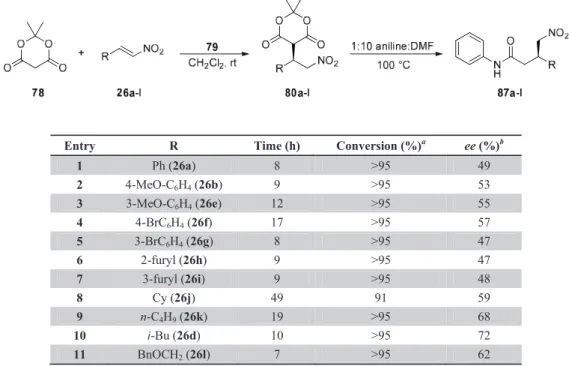
Asymmetric Organocatalytic Michael Additions
Purified by flash chromatography (50% EtOAc in hexane) and crystallized from EtOAc:hexane to yield 87e as white crystals (16 mg). Purified by flash chromatography (40% EtOAc in hexane) and crystallized from EtOH:H2O to yield 87f as white crystals (10 mg). Purified by flash chromatography (0 to 15% EtOAc in hexane) and crystallized from EtOAc:hexane to yield 87k as white crystals (9 mg).
Purified by flash chromatography (40% EtOAc in hexane) and crystallized from EtOH:H 2 O to give 87 L as white crystals (17 mg).
Total Synthesis of (–)-Hennoxazole A
The mixture was extracted with EtOAc (5 x 7 mL) and the organic phases were dried over MgSO 4 , filtered and evaporated to give the crude product as a brown oil. The mixture was extracted with EtOAc (5 x 5 mL) and the combined organic phases were dried over MgSO 4 , filtered and evaporated to give the crude product which was analyzed by 1 H NMR. The mixture was diluted with 100 mL of H2O, the phases were separated and the aqueous phase was extracted with CH2Cl2 (100 mL).
The mixture was diluted with 50 mL H2O, the phases were separated and the aqueous phase was extracted with CH2Cl2 (2 * 50 mL).

Continuous Flow Nitration of Aromatic Substrates
The reactor was set to the desired temperature and flow rate, and then 1 mL of both stock solutions were injected into the reactor via injection ports equipped with appropriate injection loops. The start of the reaction was observed by a color change to yellow either in the beaker after the end of collection, immediately after the reaction stream left the bpr, or in the reactor coil. The reactor was charged to the desired temperature and flow rate, and then the stock solutions were pumped into the reactor.
The resulting reacted mixture was collected in a separate beaker partially filled with H2O. The effluent was analyzed by HPLC by collecting the mixture in a 10 mL volumetric flask for a few seconds and then diluting to the indicated volume with 1:1 H2O:MeCN.