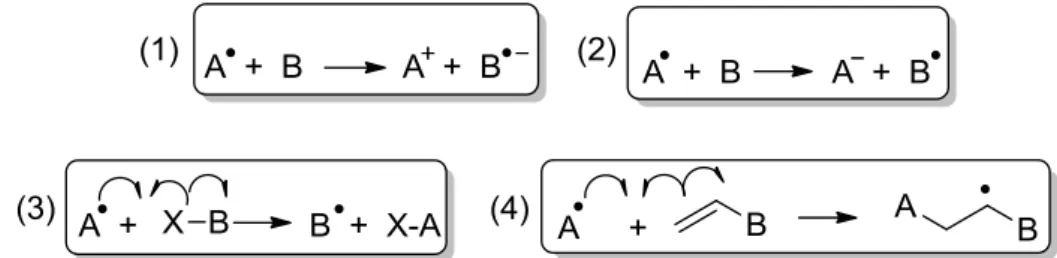
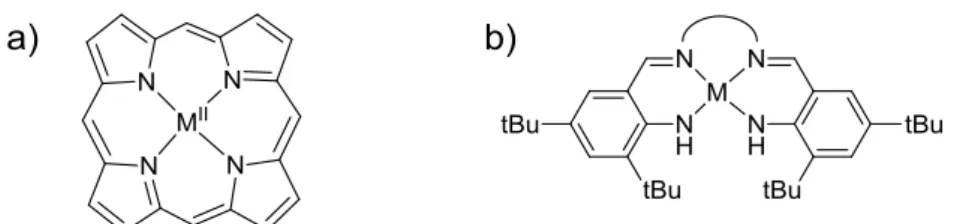
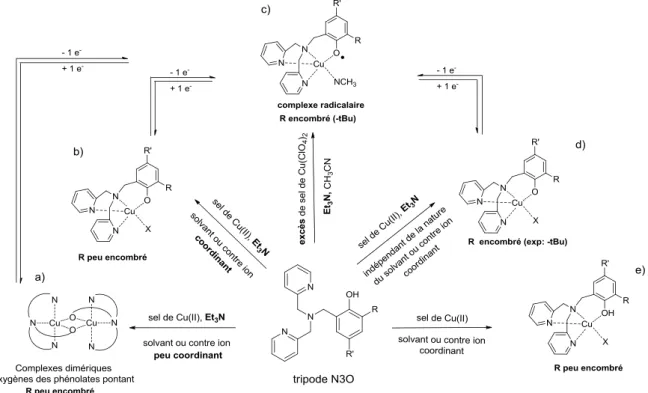
Les complexes de cuivre ont été largement décrits comme modèles de l’entité radicalaire du cuivre dans GO. De plus, le complexe [CuII(18)•]+ est silencieux en RPE en raison d'un couplage magnétique entre les spins du radical et l'ion métallique. D'autres complexes salens ont été synthétisés dans le but de moduler la flexibilité du pont et la géométrie autour de l'ion métallique.
Introduction bibliographique
- De la découverte de radicaux organiques libres par Moses Gomberg à la chimie
- Qu’est ce qu’un radical organique libre?
- La découverte capitale de Gomberg
- Un héritage de grande envergure
- Les radicaux dans les protéines
- Les radicaux, des espèces ubiquitaires
- La Galactose oxydase, une métalloenzyme unique
- Caractérisation et propriétés des radicaux phénoxyles
- Structure électronique et stabilisation
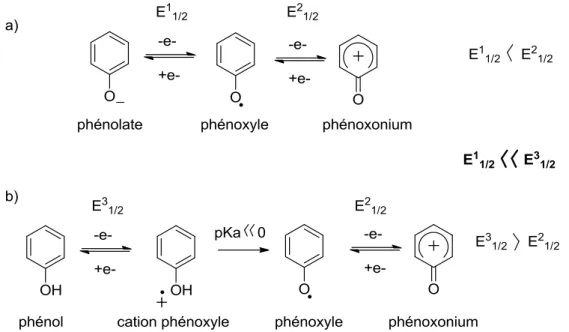
- Electrochimie des radicaux phénoxyles
- Structure de radicaux phénoxyles non coordinés
- Spectroscopie UV-Vis
- Spectroscopie RPE
- Elaboration de complexes métal-radical bioinspirés de la Galactose Oxydase
- Complexes dérivés de ligands tripodes
- Complexes dérivés du macrocycle TACN (1,4,7-triazacyclononane)
- Complexes d’o-catécholate, d’o-phenylènediimide, d’o-benzènedithiolate, d’o-
- Ligands salen (ou bases de Schiff)
- Complexes salen
- Structure générale et chimie de coordination des ligands salen
- Synthèse des ligands et complexes salen
- Réactivité de complexes salen
- Chimie radicalaire des complexes salen
Dans le cas de [NiII(1NMe2)•]+ légère augmentation de la localisation du radical sur le ligand (cycle A, 95.7. L'analyse de la densité de spin confirme le fait que le site d'oxydation est situé sur le 4-N,N- cycle diméthylamino-2-tert-butylphénolate dans le composé [Ni(3)•]+ (Figure 76-a).Ces résultats montrent donc que l'oxydation du complexe [Ni(5N4tBu . )] •+.SbF6- est centrée sur le ligand avec formation d'un radical anilinyle (type NiIII-• NH), délocalisé sur deux cycles de la molécule.
Dans le cas de [Ni(1N4tBu)]•+ et [Ni(4N4tBu)]•+, les calculs DFT prédisent une contraction symétrique de la sphère de coordination. Dans le cas du complexe [Cu(6)], l'onde monoélectronique réductrice devient également réversible, mais cette fois elle est significativement décalée vers les potentiels les plus élevés (EV). Les potentiels d'oxydation et de réduction des complexes [Ni(10)•] et [Cu(10)•] ne dépendent guère de la nature du métal, ce qui suggère que ces processus sont principalement centrés sur le ligand.
Les complexes [Ni(10)]+ et [Cu(10)]+ générés par oxydation chimique sont stables en solution plusieurs heures (durée de mesure) dans le dichlorométhane. Dans ces complexes [M(10)•], les transitions observées dans le proche IR sont des transferts de charge pyrrophénoxyle → dipyrrométhène.
Complexes radicalaires de cuivre et nickel
Influence de la fonctionnalisation en para des phénolates sur la structure
- Introduction
- Synthèse des ligands et complexes [Ni II (1 tBu )], [Ni II (1 OMe )] et [Ni II (1 NMe2 )]
- Electrochimie de [Ni II (1 tBu )], [Ni II (1 tBu )] et [Ni II (1 NMe2 )]
- Caractérisation par calculs théoriques des complexes neutres et monooxydés
- RPE de [Ni II (1 tBu ) • ] + , [Ni II (1 OMe ) • ] + et [Ni II (1 NMe2 ) • ] +
- Résonance Raman (RR) des complexes neutres et monooxydés
- Spectroscopie UV-visible (UV-vis) et proche infra rouge (IR)
- Analyse par calcul théorique des transitions dans le proche IR
- Conclusion
Les calculs prédisent également un complexe asymétrique dans le cas de [NiII(1NMe2)•]+, avec un radical phénoxyle situé sur un seul noyau phénolique. Dans le cas de [NiIII(1tBu)•]+, le DS est délocalisé dans tout le ligand avec une fraction significative portée par le centre métallique Ni (> 15%) (B3LYP et CAM-B3LYP). Pour [NiII(1OMe)•]+ et [NiII(1NMe2)•]+, des longueurs de liaison typiques d'un radical phénoxyle sont observées dans le cycle A (distribution quinoïdale).
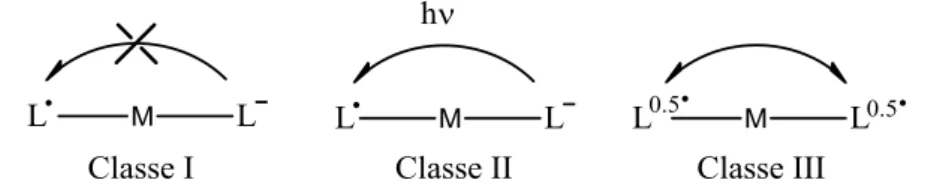
Ces résultats RPE fournissent donc des preuves expérimentales supplémentaires de la nature localisée du radical dans un seul cycle dans le complexe [NiIII(1NMe2)•]+. L'analyse de l'énergie et de la forme des bandes dans le proche IR (intensité et largeur à mi-hauteur) permet d'évaluer le degré de délocalisation dans ces systèmes à valence mixte. Il peut donc être décrit comme un radical complètement délocalisé sur les deux anneaux phénoliques du ligand, ce qui confirme les analyses précédentes (Tableau 7).125 Le composé [NiIII(1OMe)•]+ présente une bande proche IR plus large (Δν1/ 2) . = 1250 cm-1) et modérément intense (ε = 12800 cm-1).
La température dans le composé [NiII(1NMe2)•]+ n'affecte ni l'intensité ni la largeur de la bande proche IR, qui est donc décrite comme un composé de classe II à valence mixte. Dans le cas du complexe [NiII(1tBu)•]+, les orbitales donneur (β-HOMO) et accepteur (β-LUMO) correspondent au système π délocalisé sur deux noyaux aromatiques (Figure 56-a).125 En comparaison , dans les complexes [NiII( 1OMe)•]+ et [NiII(1NMe2)•]+ l'orbitale donneuse est située sur un seul anneau phénolique, et l'orbitale acceptrice correspond au deuxième anneau phénolique (Fig. 56-b et - contre).
Activité redox centrée sur le pont phénylènediamine dans les complexes
- Introduction
- Synthèse des ligands et complexes [Cu(2 OMe )] et [Cu( OMe 2)]
- Caractérisation structurale des complexes neutres
- Electrochimie de [Cu(2 OMe )] et [Cu( OMe 2)]
- Caractérisation structurale de [Cu( OMe 2)] •+ .SbF 6 -
- Caractérisation par calculs théoriques des complexes neutres et monooxydés
- Analyse par calcul théorique des transitions dans le proche IR
- Résonance Raman (RR) des complexes neutres et monooxydés
- Conclusion
La voltammétrie cyclique du complexe [Cu(2OMe)] est similaire à celle du composé [Cu(OMe2)] et présente deux vagues d'oxydation réversibles à E1/21. Ces résultats indiquent donc que le site d'oxydation est identique dans les composés [Cu(2OMe)] et [Cu(2tBu)] et centré sur le phénolate (s'oxyde en phénoxyle). Nous observons expérimentalement exactement l’effet inverse, suggérant que le site d’oxydation est différent dans ces deux molécules.
Si l'oxydation est focalisée sur le métal (le complexe diamagnétique CuIII-bis(phénolate)), les calculs prédisent une contraction de la sphère de coordination d'environ 0,04 Å, ce qui est en contradiction avec les résultats expérimentaux. l'oxydation est focalisée sur le ligand (complexe paramagnétique), cela ne devrait pas provoquer de contraction significative des liaisons Cu-O et Cu-N, ce qui reproduit les résultats expérimentaux de diffraction des rayons X. L'absence de contribution du cycle phénylènediamine indique que l'oxydation du complexe [Cu(2OMe )] conduit à un radical.
La bande à 1542 cm-1 peut être attribuée à la vibration ν8a du radical diiminobenzène,163 alors que la bande typique du radical phénoxyle n'est pas observée pour ce composé.119 Ces résultats confirment que l'oxydation dans le composé [Cu (OMe2) •]+ est centré sur le pont avec une délocalisation partielle du radical sur les orbitales phénolates. En effet, l'oxydation des complexes [Cu(OMe2)] et [Cu(2OMe)] est dans chaque cas centrée sur le ligand, mais la nature du radical est différente.
![Figure 58: Structures cristallographiques des complexes a) [Cu( OMe 2)] et b) [Cu(2 OMe )] (ellipsoïdes à 30 %)](https://thumb-eu.123doks.com/thumbv2/1bibliocom/464385.69897/99.892.133.773.528.741/figure-structures-cristallographiques-complexes-cu-ome-ome-ellipsoïdes.webp)
Complexes dissymétriques Ni(II)-salophen : déplacement du site d’oxydation
- Introduction
- Synthèse des ligands et complexes [Ni(3H)] + et [Ni(3)]
- UV-visible des complexes non oxydés [Ni(3)] et [Ni(3H)] +
- Electrochimie de [Ni(3)] et [Ni(3H)] +
- UV-visible des complexes monooxydés [Ni(3)] •+ et [Ni(3H)] •2+
- Caractérisation par RPE et calculs DFT de [Ni(3) • ] + et [Ni(3H)] •2+
- Caractérisation par calculs théoriques des complexes monooxydés
- Conclusion
Dans le cas du composé protoné [Ni(3H)]+ l'onde d'oxydation est fortement décalée vers les potentiels les plus élevés E1/21. Le spectre UV-visible du complexe monoradicalaire [Ni(2NMe2)•]+ présente des propriétés similaires (630 nm, 7890 M-1 cm-1),121 ce qui confirme que le site d'oxydation dans le complexe [Ni(3)• ] + est situé sur le cycle N,N-diméthylaminophénolate. Il est à noter que la similitude des spectres RPE de [Ni(3H)•]2+ et [Ni(2tBu)•]+ suggère que soit le radical est localisé sur un seul cycle aromatique de ce dernier complexe, soit cela.
Ligne continue : spectre expérimental, ligne pointillée : simulation réalisée à partir des paramètres donnés dans le texte. De plus, l'analyse des longueurs de liaison montre une distorsion quinoïde, localisée cette fois dans le cycle di-tert-butylé. Ceci montre que si le SOMO est majoritairement porté par le ligand dans le cas du complexe [Ni(3)•]+, la participation de l'orbitale métallique n'est pas négligeable dans le complexe [Ni(3H)•]2+.
Une telle différence est attribuée à la délocalisation supplémentaire du radical sur le groupe N,N-diméthylamino dans le cas de [Ni(3)·]+. La conséquence directe de l'augmentation de la participation du métal dans SOMO à [Ni(3H)·]2+ est l'augmentation de la valeur giso qui est théoriquement prédite et observée expérimentalement.
![Figure 69: Synthèse de [Ni(3H)] + .](https://thumb-eu.123doks.com/thumbv2/1bibliocom/464385.69897/109.892.189.727.109.281/figure-69-synthèse-de-ni-3h.webp)
Complexes salen tétra-azotés de Cuivre et de Nickel
- Introduction
- Synthèse des ligands et complexes [M(1 N4 tBu )] et [M(4 N4 tBu )]
- Caractérisation structurale de [Cu(1 N4 tBu
- Caractérisation spectroscopique des complexes neutres en solution
- Complexes de nickel [Ni(1 N4 tBu
- Complexes de cuivre [Cu(1 N4 tBu
- Electrochimie de [Cu(1 N4 tBu )], [Ni(1 N4 tBu )], [Cu(4 N4 tBu )] et [Ni(4 N4 tBu )]
- Caractérisation structurale des complexes monooxydés
- Caractérisation par calculs théoriques des complexes neutres et monooxydés
- RPE de [Ni(1 N4 tBu ) • ] + , [Cu(1 N4 tBu ) • ] + , [Ni(4 N4 tBu ) • ] + et [Cu(4 N4 tBu ) • ] +
- UV-visible et proche infra rouge (IR) des complexes monooxydés
- Analyse par calcul théorique des transitions dans le proche IR
- Conclusion
Le remplacement des phénolates par des phénylamides induit donc généralement une extension de la sphère de coordination du métal d'env. 0,05 Å. L'ajout d'un autre acide équivalent (Figure 87-b) provoque une diminution plus importante de l'intensité de la transition LMCT. Le titrage du complexe [Cu(4N4tBu. )] (Annexe 3) dans les mêmes conditions provoque également une diminution de la bande à 430 nm.
Cela peut être dû à une répartition du radical anilinyle sur les deux moitiés de la molécule. Il en est de même lorsque l'oxydation a lieu au niveau du ligand (CuII-phénoxyle) avec le radical complètement délocalisé sur les deux noyaux aromatiques de la molécule. Nos résultats expérimentaux montrent que la contribution de la forme A est plus importante dans les radicaux anilinyle que dans les phénoxyles.
Ces calculs prédisent une structure légèrement asymétrique avec une contraction de la sphère de coordination par rapport à celle du précurseur neutre [Cu(1N4tBu . )] ce qui n'est pas en accord avec les résultats expérimentaux. Ces résultats montrent donc que le poids de la forme A (Figure 78) est plus important dans les radicaux anilinyles coordonnés par rapport aux phénoxyles.
Complexes de thiosemicarbazones
- Complexes de thiosemicarbazones symétriques
- Introduction
- Synthèse des complexes [Cu(6)] et [Ni(6)]
- Electrochimie de [Cu(6)] et [Ni(6)]
- UV-vis des complexes neutres, monooxydés et réduits
- RPE des complexes neutres, monooxydés et réduits
- Conclusion
- Complexes hybrides phénol-thiosemicarbazone
- Introduction
- Synthèse des ligands et complexes Ni(7)], [Ni(8)] et [Ni(9)]
- Caractérisation structurale des complexes neutres [Ni(7)], [Ni(8)] et [Ni(9)] . 143
- UV-vis et proche IR de [Ni(7)], [Ni(8)], [Ni(7) • ] + , [Ni(7) • ] - et [Ni(8) • ] -
- RPE de [Ni(7) • ] + , [Ni(8) • ] + , [Ni(7) • ] - et [Ni(8) • ] -
- Conclusion
Les longueurs des liaisons de coordination sont plus courtes dans le cas du complexe NiIII en raison de son rayon ionique plus faible. Ceci s'explique par la déprotonation du ligand dans le complexe [Ni(6)], qui est alors enrichi en électrons et donc plus difficile à réduire. Le complexe [Cu(6)] est donc plus facile à réduire que le ligand, ce qui suggère que la réduction sera probablement centrée sur le métal dans le cas du complexe CuII.
Cette technique spectroscopique n'est donc pas adaptée pour distinguer le site d'oxydation ou de réduction dans le cas des complexes de cuivre. Bien que la réduction soit centrée sur le métal dans le cas de [CuI(6)]-, les orbitales du ligand et plus particulièrement le pont α-diimine contribuent certainement à la stabilisation du complexe. Le comportement électrochimique des complexes [Ni(7)], [Ni(8)] et [Ni(9)] a été étudié par voltamétrie cyclique dans le dichlorométhane anhydre et le THF (Figure 116) et les valeurs des différents potentiels rédox sont notés dans le tableau 16 .
La première vague d'oxydation est attribuée à la formation d'un radical phénoxyle dans le cas de [Ni(7)] (NiII-•OPh) ou thiyle dans le cas de [Ni(8)] (NiII-•SPh) . Le groupe thiyle formé par un thiophénolate dans le complexe [Ni(8)•]+ ne s'est pas révélé très stable.
![Figure 102: Structures cristallographiques de a) [Cu(6)] et b) [Ni(6)] (ellipsoïdes à 30 %)](https://thumb-eu.123doks.com/thumbv2/1bibliocom/464385.69897/139.892.133.771.416.567/figure-102-structures-cristallographiques-cu-6-ellipsoïdes-30.webp)
Complexes hybrides dipyrrométhène-phénolates de cuivre et nickel
- Introduction
- Chimie redox des porphyrines
- Ligands dipyrrométhènes
- Ligand hybride bis(di-tert-butyl-phénol)-dipyrrine
- Synthèse du ligand H 3 10 et des complexes de Cu II et de Ni II
- Caractérisation structurales des complexes neutres et calculs DFT
- Electrochimie de [Ni(10) • ] et [Cu(10) • ]
- Caractérisation structurale des complexes dioxydés [Ni(10)] + et [Cu(10)] + et calculs
- Caractérisation RPE des composés [Ni(10) • ], [Ni(10)] + , [Cu(10) • ] et [Cu(10)] +
- UV-vis et proche infra rouge de [Ni(10) • ], [Ni(10)] + , [Cu(10) • ] et [Cu(10)] + et
- Conclusion
D'autre part, le fragment dipyrrométhène devrait également favoriser l'activité rédox centrée sur le ligand. Aucune molécule exogène n'est coordonnée en position axiale par rapport au métal, ce qui suggère que l'oxydation est centrée sur le ligand [NiIII(10)•] plutôt que sur le métal [NiIII(10)]. Les résultats des calculs théoriques sont en accord avec les résultats expérimentaux et montrent une oxydation avec le centre du ligand.
Par conséquent, le complexe de cuivre se comporte de manière identique au complexe de nickel, ce qui indique que la seconde oxydation est également centrée sur le ligand. Comme dans le cas du complexe du nickel, le ligand peut être considéré comme un système à coque fermée (ligand diamagnétique, [Cu(10)ox]+) ou un système à coque ouverte (ligand diradical, [Cu(10)• • ]+ ) (Figure 134). Ces résultats suggèrent donc que les transferts de charges impliquent principalement le ligand (LLCT) plutôt que le métal.
Dans ce travail, nous avons démontré pour la première fois une chimie oxydative centrée sur le ligand dans les complexes bis(di-tert-butyl-phénol)-dipyrrine ( Figure 144 ). Les complexes de radicaux monoxyde et dioxyde avec le ligand dans une configuration à coque fermée sont extrêmement accessibles en raison de la nature trianionique du ligand précurseur déprotoné.
![Figure 123: Représentation schématique des différents états redox accessibles des complexes [M II (TPP)] (M = Ni ou Cu)](https://thumb-eu.123doks.com/thumbv2/1bibliocom/464385.69897/161.892.112.782.442.603/figure-représentation-schématique-états-redox-accessibles-complexes-tpp.webp)
Complexes de cobalt(II) impliquant des ligands redox actifs
Complexes salen de Co(II)
- Synthèse des ligands et complexes [Co(1 tBu )], [Co(1 OMe )], [Co(1 tBu )] + , [Co(1 OMe )] + ,
- Géométrie et structure électronique de [Co(1 OMe )] et [Co(1 tBu )]
- Electrochimie de [Co(1 OMe )], [Co(1 tBu )], [Co(1 OMe )(im) 2 ] + et [Co(1 tBu )(im) 2 ] +
- Caractérisation des complexes monooxydés en présence de N-méthylimidazole
- Caractérisation des complexes monooxydés dans CH 2 Cl 2
- Caractérisation des complexes dioxydés dans CH 2 Cl 2
- Conclusion
L'oxydation aérobie du complexe [Co(1OMe)] dans une solution méthanol/CH2Cl2 en présence de trois équivalents de N-méthylimidazole suivie de l'ajout de NaClO4 (qui permet au complexe de précipiter) conduit au complexe diamagnétique [Co(1OMe ) (je )2]+.C104-. Co(1tBu)] a une géométrie autour de l'ion métallique très proche de celle observée dans [Co(1OMe)], avec des longueurs de liaisons Co-O1 et Co-O2 similaires (Tableau 26). En présence de N-méthylimidazole, l'oxydation de l'espèce neutre [Co(1OMe)] en son monocation correspondant peut être réalisée par simple incubation aérobie en raison de la faible valeur de E1/21.
La diffusion de pentane dans les solutions obtenues permet d'isoler des monocristaux de [Co(1OMe)]+.SbF6-. Les atomes de base N2O2 sont coplanaires et l'ion cobalt est déplacé de 0,130 Ǻ par rapport à ce plan vers l'atome O3. Dans [Co(1OMe)]+, les longueurs de liaison Co-O sont significativement plus courtes que dans [Co(1OMe)(im)2]+ mais sont presque identiques à celles mesurées dans [Co(1OMe)] (moyenne Co-O les distances de liaison sont de 1,846(3) Ǻ dans [Co(1OMe)]).
Toutes ces données mettent en évidence la différence de structure électronique de l’ion métallique dans les deux cations [Co(1OMe)]+ et [Co(1OMe)(im)2]+. Quant à [Co(1OMe)]+, les calculs DFT mettent en évidence la contribution non négligeable de l'isomère de valence du radical Co(II) dans le cas de [Co(1tBu)]+.
Complexes salen tétra-azotés de Co(II)
- Synthèse des complexes [Co(1 N4 tBu
- Géométrie et structure électronique du complexe neutre [Co(1 N4 tBu
- Electrochimie de [Co(L 1N4 tBu
- Caractérisation du complexe monooxydé [Co(1 N4 tBu
- Caractérisation du complexe monooxydé [Co(1 N4 tBu
- Caractérisation du complexe dioxydé [Co(1 N4 tBu
- Conclusion
Complexes hybrides dipyrrométhène-phénolates de Co(II)
- Synthèse et caractérisation du complexe neutre [Co(10)]
- Caractérisation du complexe dioxydé [Co(10)] +
- Conclusion
Catalyse d'oxydation d'alcools par des comlplexes radicalaires
Introduction
- Introduction bibliographique
Catalyse d’oxydation aérobie d’alcools en aldéhydes
- Conditions opératoires
- Réactivité des complexes radicalaires pour l’oxydation aérobie de l’alcool
- Conclusion

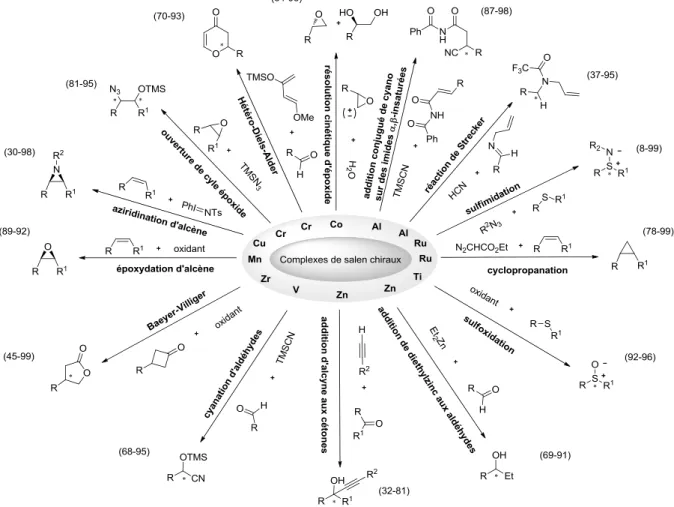
![Figure 33: Résolution cinétique par hydrolyse (HKR) d'époxydes racémiques par le complexe [Co(1 tBu )(OAc)]](https://thumb-eu.123doks.com/thumbv2/1bibliocom/464385.69897/59.892.134.760.108.335/figure-résolution-cinétique-hydrolyse-hkr-époxydes-racémiques-complexe.webp)
![Figure 41: Structure cristallographique par diffraction des rayons X de [Ni II (1 tBu ) • ] + et représentation schématique](https://thumb-eu.123doks.com/thumbv2/1bibliocom/464385.69897/69.892.160.732.105.268/figure-structure-cristallographique-diffraction-rayons-ii-représentation-schématique.webp)
![Figure 44: Représentation du complexe [Cu II (18)] (à gauche) et de sa structure cristallographique par diffraction des rayons X (à droite)](https://thumb-eu.123doks.com/thumbv2/1bibliocom/464385.69897/72.892.114.710.106.327/figure-représentation-complexe-gauche-structure-cristallographique-diffraction-rayons.webp)
