E
E
D
D
E
E
SE
S
E
NV
N
N
N
OV
O
VA
AS
S
E
E
XT
X
TR
RA
AC
CÇ
Ç
DEPARTA
V
VO
OL
LV
VI
I
M
M
S
S
F
F
AS
A
SE
ES
S
Ç
Ç
ÃO
Ã
O
S
S
OR
O
R
Fátim
UNIVERS
FACULD
AMENTO
M
ME
E
NT
N
T
O
O
,
,
O
O
P
P
OL
O
LI
I
MÉ
M
É
R
RT
TI
IV
V
A
A
E
E
M
M
a Catar
Doutoram
(Quí
SIDADE D
DADE DE C
DE QUÍM
O
O
PT
P
TI
I
MI
M
I
Z
Z
É
É
RI
R
IC
C
AS
A
S
(
(
M
M
B
B
AR
A
RR
R
rina Mo
mento em
mica Anal
2010
E LISBOA
CIÊNCIAS
ICA E BIOQ
Z
ZA
AÇ
Ç
ÃO
Ã
O
E
E
(
(P
P
OL
O
LI
IU
U
R
R
R
RA
A
D
DE
E
A
A
G
G
orais Po
m Química
ítica)
QUÍMICA
E
E
A
A
P
P
LI
L
I
CA
C
A
R
RE
E
TA
T
AN
NO
OS
S
G
G
IT
I
TA
AÇ
Ç
ÃO
Ã
O
ortugal
A
AÇ
ÇÃ
ÃO
O
D
DE
E
S
S
)
)
P
PA
AR
RA
A
O
O
(
(
S
S
B
B
S
S
E
E
E
E
E
E
)
)
E
E
D
D
E
E
SE
S
E
NV
N
N
N
OV
O
VA
AS
S
E
E
XT
X
TR
RA
AC
CÇ
Ç
DEPARTA
V
VO
OL
LV
VI
I
M
M
S
S
F
F
AS
A
SE
ES
S
Ç
Ç
ÃO
Ã
O
S
S
OR
O
R
Fátim
Profess
Depar
UNIVERS
FACULD
AMENTO
M
ME
E
NT
N
T
O
O
,
,
O
O
P
P
OL
O
LI
I
MÉ
M
É
R
RT
TI
IV
V
A
A
E
E
M
M
a Catar
Tese
sor Douto
rtamento de
Doutoram
(Quí
SIDADE D
DADE DE C
DE QUÍM
O
O
PT
P
TI
I
MI
M
I
Z
Z
É
É
RI
R
IC
C
AS
A
S
(
(
M
M
B
B
AR
A
RR
R
rina Mo
e Orientada
or José M
e Química e
mento em
mica Anal
2010
E LISBOA
CIÊNCIAS
ICA E BIOQ
Z
ZA
AÇ
Ç
ÃO
Ã
O
E
E
(
(P
P
OL
O
LI
IU
U
R
R
R
RA
A
D
DE
E
A
A
G
G
orais Po
por:
Manuel N
e Bioquímica
m Química
ítica)
QUÍMICA
E
E
A
A
P
P
LI
L
I
CA
C
A
R
RE
E
TA
T
AN
NO
OS
S
G
G
IT
I
TA
AÇ
Ç
ÃO
Ã
O
ortugal
Nogueira
a, FCUL
A
AÇ
ÇÃ
ÃO
O
D
DE
E
S
S
)
)
P
PA
AR
RA
A
O
O
(
(
S
S
B
B
S
S
E
E
E
E
E
E
)
)
Este trabalho recebeu o apoio da Fundação para a Ciência e Tecnologia através de uma Bolsa de Doutoramento no âmbito do POCI 2010 e FSE (SFRH/BD/24598/2005).
i
Agradecimentos
Quero agradecer ao meu orientador, o Professor José Manuel Nogueira, que me entregou a responsabilidade de desenvolver um trabalho inovador e em que talvez poucos acreditassem. Obrigada apoio ao longo do meu trabalho e por me dar a oportunidade de aprender e evoluir.
Ao Professor João Silva e ao Dr. Moisés Pinto, que me ensinaram tudo o que sei sobre os poliuretanos. Obrigada pela paciência que tiveram para as minhas dúvidas e perguntas básicas e por toda a orientação que me proporcionaram.
Queria agradecer à Fundação para a Ciência e Tecnologia, pela bolsa de doutoramento que me foi atribuída.
Agradeço também ao Dr. Pedro Vaz, pelas úteis explicações no trabalho realizado com recurso à técnica de FTIR-ATR e à Engª. Paula Branco, pelos conhecimentos transmitidos durante o estudo das hormonas esteróides.
À Carla e à Rita por terem sido óptimas amigas, ouvintes e colegas de trabalho. À Susana, à Tânia, à Ana Mestre e ao Paulo Madeira agradeço todo a amizade, o apoio e a paciência.
Ao meu irmão por me ouvires e mostrares que a vida não pode ser desperdiçada, que para tudo há uma solução!
Às pessoas que mais admiro, que tomo como exemplo e a quem mais devo tudo o que alcancei, os meus Pais, que tudo fizeram por mim. Vocês sempre me incentivaram e apoiaram incondicionalmente. Obrigada por tudo!! Adoro-vos!!
Por último, ao meu Paulo... Obrigada, Paulinho, por me dares a energia que eu mais precisava para ultrapassar a barreira de activação nos momentos mais difíceis, pela paciência que tiveste durante todo este tempo e pela ajuda em todos os momentos. Obrigada por criares entropia positiva na minha vida e fazeres dela algo melhor e em que é maravilhoso viver!
iii
Prefácio
A sociedade actual vive constantemente em busca de novas soluções que nos permitam desenvolver formas de “poupar” o ambiente, havendo na área da Química uma preocupação constante nesse sentido. No contexto das soluções ambientalmente sustentáveis nesta área, a “química verde” tem vindo a revelar‐se uma aposta ganha no que se refere à invenção, desenvolvimento e aplicação de produtos e processos químicos que reduzem ou eliminam o uso e a geração de substâncias perigosas.
Esta nova filosofia inclui doze princípios, entre os quais alguns aludem à prevenção do uso de substâncias tóxicas, o que influencia de forma directa as técnicas de preparação de amostra em química analítica, uma vez que as metodologias implementadas na maioria dos laboratórios não se encontram em sintonia com estes princípios, tendo em conta as grandes quantidades de solventes tóxicos utilizados diariamente nos vários processos de extracção mais utilizados. Também as exigências de redução do tempo analítico e de automatização no trabalho de rotina conduzem a desenvolvimentos analíticos de miniaturização, nomeadamente, na redução da quantidade de solventes e volumes de amostra.
Neste contexto, as técnicas de extracção têm vindo a ser gradualmente substituídas por técnicas solventless, i.e. que visam a redução substancial ou mesmo a eliminação do consumo de solventes orgânicos. Nesta área destacam‐se as técnicas de extracção sortiva, nomeadamente, a microextracção em fase sólida e a extracção sortiva em barra de agitação, as quais têm vindo a mostrar‐se ambientalmente favoráveis.
Nesta dissertação, estudaram‐se novas fases poliméricas à base de poliuretano para a extracção sortiva com o intuito de colmatar as dificuldades demonstradas pelas fases poliméricas à base de polidimetilsiloxano na extracção de compostos com características mais polares. Deste estudo resultaram diversas publicações em revistas científicas da especialidade, bem como apresentações orais e em painel:
Publicações em Revistas Científicas Internacionais
• Optimization of Polyurethane Foams for Enhanced Stir Bar Sorptive Extraction of
Triazinic Herbicides in Water Matrices, Fátima C.M. Portugal, Moisés L. Pinto, J.M.F.
iv
• Potentialities of Polyurethane Foams for Trace Level Analysis of Triazinic
Metabolites in Water Matrices by Stir Bar Sorptive Extraction, Fátima C.M. Portugal,
Moisés L. Pinto, João Pires, J.M.F. Nogueira, J. Chromatogr. A 1217 (2010) 3707. • Static Headspace Analysis using Polyurethane Phases ‐ Application to coffee volatile
fraction characterization and discrimination, C. Rodrigues, Fátima C. M. Portugal, J. M.
F. Nogueira (submetido)
• Characterization of Polyurethane Foams used as Coatings in Sorptive Extraction, Fátima C.M. Portugal, Moisés L. Pinto, João Pires, J.M.F. Nogueira (em preparação) • Progress in Sample Enrichment Techniques for Trace Levels of Steroid Sex Hormones
in Aqueous Matrices, Fátima C.M. Portugal, Ana Rita M. Silva, J.M.F. Nogueira (em
preparação)
Comunicações em Painel
• New Polymeric Phases for Stir Bar Sorptive Extraction – 29th International Symposium
on Capillary Chromatography, IOPMS, 2006, Riva del Garda, Itália.
• Development of New Polymeric Phases for Stir Bar Sorptive Extraction and Their
Application in the Determination of Triazinic Pesticides in Water Matrices – XX
Encontro Nacional da SPQ, 2006, FCT‐UNL/SPQ, Lisboa, Portugal.
• Application of New Polymeric Phases for Stir Bar Sorptive Extraction in the Analysis
of Triazinic Herbicides – 6º Encontro Nacional da Divisão de Química Analítica da SPQ,
2007, IST/SPQ, Lisboa, Portugal.
• New Polymeric Phases for Stir Bar Sorptive Extraction in the Analysis of Endocrine
Disrupting Chemicals in Environmental Matrices – 31st International Symposium on High Performance Liquid Phase Separations and Related Techniques, IOPMS, 2007,
Ghent, Bélgica.
• New Generation Polymers for SBSE: Characterization and Application in the Analysis
of Endocrine Disruptors – 5º Encontro Nacional de Cromatografia, UA/SPQ, 2007,
Aveiro, Portugal.
• Polyurethane Foams as New Generations Polymeric Phases for Stir Bar Sorptive
Extraction: Application in the Analysis of Triazines in Environmental Water Matrices
– 32nd International Symposium on Capillary Chromatography, IOPMS, 2008, Riva del
v
• Polyurethane Foams For Enhanced Stir Bar Sorptive Extraction – 1ª Encontro de
Jovens Químicos, IST/SPQ, 2008, Lisboa, Portugal.
• Application of Polyurethane Foams to Characterize Aroma Compounds from Coffee
Blends – 4th International Symposium on Recent Advances in Food Analysis, 2009,
Praga, República Checa. • Advantages of the Application of Polyurethane Foams for Stir Bar Sorptive Extraction of Triazinic Metabolites on Water Matrices – 6º Encontro Nacional de Cromatografia, UMa/SPQ, 2009, Funchal, Portugal. • Application of HSSE(PU) for the Analysis of Aroma Compounds in Commercial Coffees – 6º Encontro Nacional de Cromatografia, UMa/SPQ, 2009, Funchal, Portugal. • Novas Estratégias Analíticas para Determinação de Hormonas Sexuais Esteróides em Matrizes Aquosas – 6º Encontro Nacional de Cromatografia, UMa/SPQ, 2009, Funchal, Portugal. • Optimização do Método de Ensaio para Análise de Hormonas Esteróides Sexuais em
Amostras Ambientais por LC‐(ESI)MS/MS – 6º Encontro Nacional de Cromatografia,
UMa/SPQ, 2009, Funchal, Portugal.
• Analysis of Aroma Compounds in Gourmet Coffees by HSSE(PU) – 6º Encontro
Nacional de Cromatografia, UMa/SPQ, 2009, Funchal, Portugal.
Comunicações Orais
• Há mais vida para além do PDMS – Novos Materiais Poliméricos para Extracção
Sorptiva de Compostos Polares – Fátima Portugal, Seminários do Centro de Química e
Bioquímica da Faculdade de Ciências da Universidade de Lisboa, 31 de Março de 2009
vii
Resumo
O presente trabalho propõe a síntese, o desenvolvimento, a optimização e a caracterização de novas fases poliméricas à base de poliuretanos (PU) para aplicação em técnicas de extracção sortiva.
Numa primeira fase, procedeu‐se à optimização da formulação das espumas de PU, tendo em conta o seu melhor desempenho na extracção sortiva em barra de agitação (SBSE), incluindo a variação das quantidades de óleo de silicone e de água e a adição de diferentes polióis. A formulação optimizada (P20) foi posteriormente caracterizada de modo a avaliar a sua estrutura, morfologia, estabilidade e resistência térmica, mecânica e química, recorrendo a técnicas de espectroscopia, microscopia e termogravimetria, entre outras. A configuração do formato de P20 foi igualmente optimizada, assim como as condições de condicionamento e regeneração, com vista à sua reutilização.
Do ponto de vista analítico, combinou‐se a SBSE(P20) com técnicas cromatográficas para análise de compostos modelo (herbicidas triazínicos, metabolitos triazínicos e hormonas esteróides sexuais) em fase líquida, os quais são considerados prioritários no ambiente aquático. Neste contexto, realizaram‐se ensaios em amostras aquosas fortificadas, sob condições instrumentais e experimentais optimizadas, tendo‐se obtido um bom desempenho, com eficiência e reprodutibilidade convenientes, gamas de trabalho adequadas, boa selectividade e sensibilidade, bem como recuperações aceitáveis. A aplicação das metodologias optimizadas a matrizes aquosas ambientais demonstrou igualmente bom desempenho. Por comparação da SBSE(P20) com as barras comerciais de SBSE contendo polidimetilsiloxano (PDMS), provou‐se que a primeira evidencia uma eficiência analítica mais elevada e maior abrangência para monitorizar compostos alvo numa gama de polaridades alargada.
A formulação P20 optimizada foi ainda aplicada em fase gasosa, na análise de compostos voláteis modelo (aroma do café) no espaço de cabeça, HSSE(P20). Este estudo demonstrou que esta nova fase polimérica evidencia, de igual modo, a abrangência e o bom desempenho apresentados em fase líquida, comparativamente a outras técnicas de extracção sortiva.
Dos dados obtidos, concluiu‐se que foi possível alcançar os objectivos previamente delineados, nomeadamente, a síntese de uma nova fase polimérica para SBSE, demonstrando‐ se a grande capacidade extractiva dos PUs como fases poliméricas alternativas para implementação em técnicas de extracção sortiva.
viii
P
ALAVRASC
HAVETécnicas de extracção sortiva; SBSE; HSSE; Espumas de Poliuretano (PU); Novas fases poliméricas; Triazinas; Metabolitos; Hormonas; Aroma do café.
ix
Abstract
This work aims the synthesis, development, optimization and characterization of new polymeric phases based on polyurethanes foams (PU) for application in sorptive extraction techniques.
In a first step, we performed the optimization of the PU formulation, taking into account its best performance in stir bar sorptive extraction (SBSE), including the variation of the silicone oil and water contents and the addition of different polyols. The optimized formulation (P20) was subsequently characterized in order to assess its structure, morphology, stability and thermal, mechanic and chemical resistance, using spectroscopic, microscopic and thermogravimetric techniques, among others. The geometric configuration of P20 was also optimized, as well as the conditioning and regeneration procedures, in order to assure the reuse of the bars.
From an analytical point of view, SBSE(P20) was combined with chromatographic techniques for the analysis of model compounds (triazinic herbicides, triazinic metabolites and steroid sexual hormones) in liquid phase, which are considered prioritary in the aquatic environment. In this context, assays were performed in spiked water samples, under instrumental and experimental optimized conditions, with a good performance, convenient efficiency and reproducibility, suitable linearity, good selectivity and sensitivity, as well as acceptable recoveries. The application of the optimized methodologies to environmental water samples has shown good performance. The comparison of SBSE(P20) with the commercial stir bars with polydimethylsiloxane (PDMS) for SBSE proved that the former has higher analytical efficiency, as well as a high coverage to monitor target compounds in a wide range of polarities.
The optimized P20 formulation was also applied in the gas phase, in the analysis of volatile model compounds (coffee aroma) in the headspace, HSSE(P20). This study has shown that this new polymeric phase evidences the wide range and the good performance obtained in liquid phase, comparatively to other sorptive extraction techniques.
From the data obtained, we can conclude that it was possible to achieve the previously defined goals, namely, the synthesis of a new polymeric phase for SBSE, proving the great extractive ability of the PUs as alternative polymeric phases for implementation in sorptive extraction techniques.
x
K
EYWORDSSorptive extraction techniques; SBSE; HSSE; Polyurethane foams (PU); Novel polymeric phases; Triazines; Metabolites; Hormones; Coffee Aroma.
xi
Índice Geral
Agradecimentos i Prefácio iii Resumo vii Palavras‐Chave viii Abstract ix Keywords x Índice Geral xi Índice de Figuras xvii Índice de Tabelas xxiv Abreviaturas xxvii Capítulo 1 – Técnicas de Extracção Sortiva 1 1.1. Introdução Geral 3 1.2. Extracção em Fase Sólida (SPE) 3 1.3. Extracção Sortiva 5 1.3.1. Microextracção em Fase Sólida (SPME) 6 1.3.2. Extracção Sortiva em Barra de Agitação (SBSE) 8 1.3.2.1. Conceitos teóricos 9 1.3.2.2. Desenvolvimento do método experimental 11 1.3.2.3. Novas fases poliméricas 13 1.4. Referências Bibliográficas 15 Capítulo 2 ‐ Espumas de Poliuretano 17 2.1. Introdução Geral 19 2.2. Química dos PUs 20 2.2.1. Isocianatos 21 2.2.1.1. Diisocianato de tolueno (TDI) 23xii 2.2.1.2. 4,4’‐difenilmetano diisocianato (MDI) 23 2.2.2. Polióis 24 2.2.3. Agentes de Expansão 25 2.2.4. Catalisador 26 2.2.5. Agente Nucleador ou Tensioactivo 27 2.3. Propriedades das Espumas de PU 28 2.3.1. Massa Específica Aparente 28 2.3.2. Volume de Células Abertas 28 2.3.3. Adsorção Física de Gases e Vapores 28 2.4. Espumas de PU Flexíveis 29 2.5. Aplicações Analíticas dos PUs 30 2.6. Referências Bibliográficas 31 Capítulo 3 – Compostos Modelo para Aplicação Analítica dos PUs 33 3.1. Introdução 35 3.2. Aplicações em fase líquida 35 3.1.1. Desreguladores Endócrinos 35 3.1.1.2. Herbicidas triazínicos e seus metabolitos 38 3.1.1.2. Hormonas esteróides 42 3.3. Aplicações em fase gasosa 45 3.4. Referências Bibliográficas 48 Capítulo 4 – Técnicas Cromatográficas 53 4.1. Perspectiva Histórica 55 4.2. Processo Cromatográfico e Conceitos Teóricos 55 4.3. Cromatografia Gasosa (GC) 59 4.3.1. Gás de Arraste 60 4.3.2. Sistema de Injecção 61 4.3.3. Coluna Cromatográfica 62 4.3.4. Sistema de Detecção 64
xiii 4.3.4.1. Cromatografia Gasosa – Espectrometria de Massa (GC‐MS) 64 4.3.5. Sistema de Aquisição e Tratamento de Dados 67 4.4. Cromatografia Líquida de Alta Eficiência (HPLC) 68 4.4.1. Fase Móvel 69 4.4.2. Sistema de Injecção 70 4.4.3. Coluna Cromatográfica 71 4.4.4. Sistema de Detecção 72 4.4.5. Sistema de Aquisição e Tratamento de Dados 73 4.5. Referências Bibliográficas 74 Capítulo 5 – Técnicas Microscópicas e Espectroscópicas 75 5.1. Microscopia Electrónica de Varrimento (SEM) 77 5.2. Espectroscopia de Infravermelho com Transformada de Fourier (FTIR) 78 5.3. Referências Bibliográficas 81 Capítulo 6 – Procedimentos Experimentais 83 6.1. Síntese de Espumas de PU 85 6.2. Determinação do Volume Externo e Massa Específica Aparente 86 6.3. Determinação da Densidade e Volume de Células Abertas 87 6.4. Termogravimetria com Calorimetria Diferencial de Varrimento (TG‐DSC) 87 6.5. Microscopia Electrónica de Varrimento (SEM) 87
6.6. Espectroscopia de Infravermelho com Transformada de Fourier (FTIR) no modo de
Reflectância Total Atenuada (ATR) 88 6.7. Isotérmicas de Adsorção de Furano 88 6.8. Preparação de Barras de PU para Ensaios de SBSE 89 6.9. Aplicação de Espumas de PU em Ensaios de SBSE(PU)‐LD 90 6.9.1. Ensaios por SBSE(PU)‐LD 90 6.9.2. Ensaios por HSSE(PU)‐LD 91 6.9.3. Ensaios de SBSE(PDMS)‐LD e HSSE(PDMS)‐LD 91 6.10. Análise Cromatográfica 92
xiv 6.10.1. HPLC‐DAD 92 6.10.2. LVI‐GC‐MS 93 6.11. Solventes e Padrões 94 6.12. Preparação de Soluções 96 6.12.1. Solventes para Fase Móvel 96 6.12.2. Soluções dos Analitos Estudados 96 6.13. Outro Material e Equipamento 97 6.14. Referências Bibliográficas 98 Capítulo 7 – Síntese, Optimização e Caracterização de Espumas de PU 99 7.1. Introdução 101 7.2. Apresentação e Discussão de Resultados 101 7.2.1. Desenvolvimento da Formulação da Espuma de PU 102 7.2.1.1. Variação das quantidades de água e óleo de silicone 103 7.2.1.2. Introdução de diferentes polióis 104 7.2.2. Relação entre % de MDI, Massa Específica Aparente e Recuperação Média de ATZ 108 7.2.3. Caracterização de P20 112 7.2.3.1. Determinação da densidade, volume de células abertas e conteúdo em ligações ureia de P20 112 7.2.3.2. Espectroscopia de Infravermelho com Transformada de Fourier (FTIR) 113 7.2.3.3. Termogravimetria com calorimetria diferencial de varrimento (TG‐DSC) 115 7.2.3.4. Microscopia electrónica de varrimento (SEM) 116 7.2.4. Estudo do Condicionamento e Regeneração de P20 117 7.2.5. Influência do Número de Células Abertas e da % de Ligações Ureia 122 7.2.6. Avaliação da Melhor Geometria de P20 para a Extracção 125 7.2.7. Tratamento de P20 com Ácidos e Bases Fortes 127 7.2.8. Tratamento de P20 com Solventes Orgânicos 129 7.2.9. Estudos de Adsorção de Furano 131 7.3. Conclusões 132 7.4. Referências Bibliográficas 133
xv Capítulo 8 – SBSE(P20)‐LD/HPLC‐DAD de Herbicidas Triazínicos 135 8.1. Introdução 137 8.2. Apresentação e Discussão de Resultados 138 8.2.1. Optimização das Condições Cromatográficas em HPLC‐DAD 138 8.2.2. Optimização das Condições de Extracção por SBSE(P20)‐LD 140 8.2.2.1. Condições de LD 141 8.2.2.2. Condições de SBSE(P20) 144 8.2.3. Validação da Metodologia SBSE(P20)‐LD/HPLC‐DAD 147 8.2.4. Comparação entre SBSE(P20) e SBSE(PDMS) 149 8.2.5. Aplicação a Matrizes Reais 152 8.3. Conclusões 155 8.4. Referências Bibliográficas 155 Capítulo 9 – SBSE(P20)‐LD/HPLC‐DAD de Metabolitos Triazínicos 157 9.1. Introdução 159 9.2. Apresentação e Discussão de Resultados 161 9.2.1. Optimização das Condições Cromatográficas em HPLC‐DAD 161 9.2.2. Optimização das Condições de Extracção por SBSE(P20)‐LD 163 9.2.2.1. Condições de LD 163 9.2.2.2. Condições de SBSE(P20) 166 9.2.3. Validação da Metodologia SBSE(P20)‐LD/HPLC‐DAD 172 9.2.4. Comparação entre SBSE(P20) e SBSE(PDMS) 173 9.2.5. Aplicação a Matrizes Reais 175 9.3. Conclusões 177 9.4. Referências Bibliográficas 177 Capítulo 10 – SBSE(P20)‐LD/HPLC‐DAD de Hormonas Esteróides 179 10.1. Introdução 181 10.2. Apresentação e Discussão de Resultados 181
xvi 10.2.1. Condições Cromatográficas do Sistema HPLC‐DAD 182 10.2.2. Optimização das condições de extracção por SBSE(P20)‐LD 182 10.2.2.1. Condições de LD 183 10.2.2.2. Condições de SBSE(P20) 186 10.2.3. Comparação entre SBSE(P20) e SBSE(PDMS) 190 10.3. Conclusões 192 10.4. Referências Bibliográficas 192 Capítulo 11 – HSSE(P20)‐LD/LVI‐GC‐MS de Compostos Voláteis 195 11.1. Introdução 197 11.2. Apresentação e Discussão de Resultados 197 11.2.1. Optimização das Condições de Extracção por HSSE(P20)‐LD 197 11.2.1.1. Condições de LD 198 11.2.1.2. Condições de HSSE(P20) 199 11.2.2. Comparação entre HSSE(P20), HSSE(PDMS) e HS‐SPME(CAR/PDMS) 201 11.3. Conclusões 207 11.4. Referências Bibliográficas 207 Capítulo 12 – Conclusões Gerais e Perspectivas Futuras 209 Anexo 1 – Curvas de Calibração Obtidas no Estudo de Herbicidas Triazínicos I Anexo 2 – Curvas de Calibração Obtidas no Estudo de Metabolitos Triazínicos VII
xvii
Índice de Figuras
Figura 1.1 – Representação genérica dos principais passos analíticos em SPE. 4
Figura 1.2 – Representação esquemática de uma seringa de SPME durante extracção e
dessorção do composto num cromatógrafo gasoso. 6
Figura 1.3 – Representação esquemática de uma barra de SBSE (a) e durante o processo de
extracção (b). 8
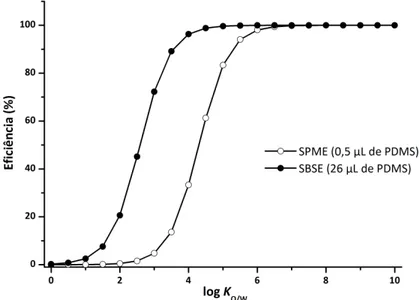
Figura 1.4 – Comparação da eficiência extractiva por SPME (0,5 μL em PDMS) e SBSE (26 μL em
PDMS) em função do log KO/W, em idênticas condições experimentais.
11
Figura 1.5 – Representação esquemática de uma barra de agitação de duas fases. 14
Figura 2.1 – Estrutura molecular do grupo uretano. 20
Figura 2.2 – Representação da reacção química entre um diisocianato e um poliol, com
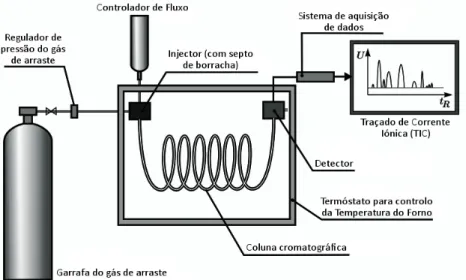
formação do grupo uretano, em que R é uma cadeia poliéter ou poliéster. 20 Figura 2.3 – Estruturas moleculares de ressonância que descrevem o grupo isocianato. 21 Figura 2.4 – Principais reacções químicas dos isocianatos. 22 Figura 2.5 – Isómeros do TDI. 23 Figura 2.6 – Estruturas moleculares dos vários isómeros do MDI. 24 Figura 2.7 – Formação do grupo ureia com libertação de CO2. 26 Figura 3.1 – Estrutura molecular geral de uma s‐triazina. 39 Figura 3.2 – Proposta dos metabolitos mais comuns de s‐triazinas e relações correspondentes com os compostos de partida. 41 Figura 3.3 – Estruturas moleculares de algumas hormonas esteróides. 44 Figura 3.4 – Estruturas moleculares de alguns compostos normalmente encontrados no aroma do café. 47 Figura 4.1 – Cromatograma típico. 56 Figura 4.2 – Exemplos de cromatogramas para diferentes valores de resolução. 58 Figura 4.3 – Esquema simplificado de um cromatógrafo gasoso. 59 Figura 4.4 – Curvas de van Deemter para diferentes gases de arraste. 60 Figura 4.5 – Estrutura molecular geral do polisiloxano. 63 Figura 4.6 – Esquema simplificado de um cromatógrafo líquido. 68
xviii
Figura 4.7 – Injector por sistema de loop: posição de introdução da amostra(a) e posição de
injecção da amostra no sistema (b). 71
Figura 4.8 – Estrutura molecular simplificada de uma fase estacionária quimicamente ligada às
partículas de sílica. 72
Figura 5.1 – Recolha de electrões secundários a partir de uma amostra tridimensional no
decorrer de uma experiência de SEM. 78
Figura 6.1 – Estruturas moleculares de alguns dos componentes utilizados para síntese de
espumas de PU. 86
Figura 6.2 – Diagrama da instalação manual em vidro utilizada nos ensaios de adsorção. 89
Figura 7.1 – Cromatograma obtido por HPLC‐DAD para as três triazinas estudadas; SMZ (1), ATZ
(2), TBZ (3). 101
Figura 7.2 – Imagens obtidas de uma barra de PU (a) e barras sob agitação em frascos de
amostragem com água ultra‐pura (b). 102
Figura 7.3 – Relação entre a recuperação média de ATZ (%) e a massa específica aparente
(kg/m3) para os polímeros da tabela 7.8. 110
Figura 7.4 – Relação entre a recuperação média de ATZ (%) e a massa específica aparente
(kg/m3) para os polímeros sintetizados com glicerol na formulação. 111
Figura 7.5 – Relação entre a massa específica aparente (kg/m3) e a % de MDI para os polímeros
sintetizados com glicerol na formulação. 111
Figura 7.6 – Relação entre a recuperação média de ATZ (%) e a % de MDI para os polímeros
sintetizados com glicerol na formulação. 112
Figura 7.7 – Relação entre a recuperação média de ATZ (%) e a % de glicerol da formulação. 112
Figura 7.8 – Espectro de P20 obtido por FTIR‐ATR, com resolução de 4 cm‐1e recolha de 264
varrimentos. 114
Figura 7.9 – Espectro obtido por espectroscopia de infravermelho do MDI. 114
Figura 7.10 – Termograma obtido por TG‐DSC para a formulação P20. 116
Figura 7.11 – Microfotografias SEM de alguns dos polímeros sintetizados, obtidos com uma
ampliação de 75x. 117
Figura 7.12– Cromatogramas obtidos por HPLC‐DAD dos brancos obtidos após os vários tipos de
condicionamento estudados. 118
Figura 7.13 – Recuperações médias obtidas por SBSE(P20)‐LD/HPLC‐DAD usando barras de P20
não condicionadas, condicionadas com tratamento US, extracção Soxhlet e extracção soxhlet seguida de tratamento US.
xix
Figura 7.14 – Microfotografias SEM de P20 não condicionado e condicionado com tratamento
US e extracção Soxhlet, obtidos com uma ampliação de 75x. 120
Figura 7.15 – Cromatogramas obtidos por HPLC‐DAD após ensaios de LD (20 min sob
tratamento US) de barras regeneradas com um ciclo de MeOH (a), dois ciclos de MeOH (b) e dois ciclos de ACN seguidos de dois ciclos de MeOH (c), barras não utilizadas (d) e barras utilizadas não regeneradas (e).
121
Figura 7.16 – Espectros obtidos de barras de P20 por FTIR‐ATR com resolução de 4 cm‐1 e recolha de 264 varrimentos, após condicionamento (a), após a extracção (b), após extracção e LD (c), após regeneração de P20 (d), após uma segunda extracção (e) e após segunda extracção e LD (f).
122
Figura 7.17 – Recuperações médias normalizadas ao volume de PU envolvido para espumas
com diferente número de células abertas. 124
Figura 7.18 – Perfis de extracção para diferentes geometrias de P20, tomando os compostos
modelo SMZ (a), ATZ (b) e TBZ (c) como exemplo. 125
Figura 7.19 – Recuperações médias obtidas para diferentes comprimentos da barra de P20,
tomando como exemplo os compostos modelo SMZ, ATZ e TBZ. 126
Figura 7.20 – Recuperações médias comparativas obtidas com P20 e PDMS, para volumes iguais
dos dois polímeros, tomando como exemplo os compostos modelo SMZ, ATZ e TBZ. 127
Figura 7.21 – Recuperações médias obtidas com P20 após tratamento ácido e básico, tomando
como exemplo os compostos modelo SMZ, ATZ e TBZ. 128
Figura 7.22 – Espectros obtidos por FTIR‐ATR com resolução de 4 cm‐1 e recolha de 264 varrimentos, após tratamento ácido e básico de P20 e de barras de P20 sem tratamentos desta natureza.
129
Figura 7.23 – Recuperações médias obtidas com P20 após tratamento de barras com diversos
solventes orgânicos, tomando como exemplo os compostos modelo SMZ, ATZ e TBZ. 130
Figura 7.24 – Espectros obtidos por FTIR‐ATR com resolução de 4 cm‐1 e recolha de 264 varrimentos, após tratamento de P20 com diversos solventes orgânicos e de uma barra de P20 sem tratamento. 131 Figura 7.25 – Isotérmicas de adsorção de furano numa amostra de P20. 132 Figura 8.1 – Estruturas moleculares dos sete herbicidas triazínicos estudados. 138 Figura 8.2 – Cromatograma obtido por HPLC‐DAD sob condições instrumentais optimizadas para
a mistura de triazinas estudadas; simazina (1), atrazina (2), prometon (3), ametrina (4), propazina (5), prometrina (6), terbutrina (7).
139
xx
Figura 8.3 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para as
triazinas em estudo na optimização do solvente de LD. 141
Figura 8.4 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para as
triazinas em estudo na optimização do tempo de retroextracção sob tratamento US. 142
Figura 8.5 – Cromatogramas obtidos por HPLC‐DAD de uma solução padrão dos sete herbicidas
triazínicos (a), um extracto obtido após a 1ª retroextracção (b) e um extracto obtido após a 2ª retroextracção (c).
142
Figura 8.6 – Efeito nas recuperações médias do passo de evaporação no processo de
preparação das amostras por SBSE(P20)‐LD seguido de HPLC‐DAD. 143
Figura 8.7 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para as
triazinas em estudo na optimização do tempo de extracção. 144
Figura 8.8 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para as
triazinas em estudo na optimização da velocidade de agitação. 145
Figura 8.9 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para as
triazinas em estudo na optimização da força iónica da matriz aquosa. 146
Figura 8.10 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para as
triazinas em estudo na optimização do conteúdo orgânico da matriz aquosa. 146
Figura 8.11 – Recuperações médias obtidas para as triazinas em estudo sob condições
experimentais optimizadas de acordo com a metodologia SBSE(P20)‐LD/HPLC‐DAD desenvolvida.
148
Figura 8.12 – Comparação das recuperações médias normalizadas ao volume polimérico
envolvido obtidas para os sete herbicidas usando SBSE(PDMS, 126 µL) e SBSE(P20, 71 µL), seguido de LD‐HPLC‐DAD, sob condições experimentais optimizadas.
149
Figura 8.13 – Linha teórica de equilíbrio e recuperação média obtida por HSSE(P20); simazina
(1), atrazina (2), prometon (3), ametrina (4), propazina (5), prometrina (6), terbutrina (7). 151
Figura 8.14 – Perfis cromatográficos obtidos por HPLC‐DAD para (a) a mistura padrão de
herbicidas triazínicos estudados, (b) herbicidas triazínicos em água subterrânea após SBSE(P20)‐ LD e (c) herbicidas triazínicos em água superficial após SBSE(P20)‐LD, sob condições experimentais optimizadas; simazina (1), atrazina (2), prometon (3), ametrina (4), propazina (5), prometrina (6), terbutrina (7). 154 Figura 9.1 – Estruturas moleculares dos cinco metabolitos triazínicos estudados. 159 Figura 9.2 – Formas tautoméricas da OH‐ATR a diferentes valores de pH. 160
xxi Figura 9.3 – Cromatograma obtido por HPLC‐DAD sob condições instrumentais optimizadas da mistura de metabolitos estudados a 213 nm (a) e a 225 nm (b); OH‐DEA (1), DIA (2), DEA (3), OH‐ATR (4), DTBZ (5). 162
Figura 9.4 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para os
metabolitos em estudo na optimização do solvente de LD. 164
Figura 9.5 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para os
metabolitos em estudo na optimização do tempo de retroextracção sob tratamento US. 164
Figura 9.6 – Recuperações obtidas após a 1ª retroextracção de 60 min e a 2ª retroextracção de
20 min para os metabolitos em estudo para avaliação do efeito de memória. 165
Figura 9.7 – Efeito nas recuperações médias do passo de evaporação no processo de
preparação das amostras por SBSE(P20)‐LD seguido de HPLC‐DAD. 166
Figura 9.8 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para os
metabolitos em estudo na optimização da velocidade de agitação. 166
Figura 9.9 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para os
metabolitos em estudo na optimização do tempo de extracção. 167 Figura 9.10 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para os metabolitos em estudo na optimização do pH da matriz aquosa. 168 Figura 9.11 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para os metabolitos em estudo na optimização da força iónica da matriz aquosa. 169 Figura 9.12 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para os metabolitos em estudo na optimização do conteúdo orgânico da matriz aquosa. 169 Figura 9.13 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para os
metabolitos em estudo na optimização do conteúdo orgânico e da força iónica da matriz aquosa.
170
Figura 9.14 – Recuperações médias obtidas para os metabolitos em estudo sob condições
experimentais optimizadas de acordo com a metodologia SBSE(P20)‐LD/HPLC‐DAD desenvolvida.
172
Figura 9.15 – Comparação das recuperações médias normalizadas ao volume polimérico
envolvido obtidas para os cinco metabolitos triazínicos usando SBSE(PDMS, 126 µL) e SBSE(P20, 86 µL), seguido de LD‐HPLC‐DAD, sob condições experimentais optimizadas.
173
Figura 9.16 – Perfis cromatográficos obtidos por HPLC‐DAD para a mistura de metabolitos
triazínicos estudados (a) e metabolitos em água superficial após extracção por SBSE(P20)‐LD, sob condições experimentais optimizadas (b); OH‐DEA (1), DIA (2), DEA (3), OH‐ATR (4), DTBZ (5).
xxii
Figura 10.1 –Perfil cromatográfico obtido por HPLC‐DAD de uma solução padrão das 8
hormonas em estudo; estriol (1), 19‐noretisterona (2), estrona (3), 17β‐estradiol (4), dietilstilbestrol (5), D‐(‐)‐norgestrel (6), progesterona (7), mestranol (8). 182 Figura 10.2 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para as hormonas em estudo na optimização do solvente de LD. 183 Figura 10.3 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para as hormonas em estudo na optimização do tempo de retroextracção sob tratamento US. 184 Figura 10.4 – Perfis cromatográficos obtidos por HPLC‐DAD para avaliação do efeito de memória para as hormonas em estudo a um c.d.o. de 240 nm; 2ª retroextracção de 15 min (a); padrão de controlo das oito hormonas em estudo (b); 1ª retroextracção de 45 min (c). 184
Figura 10.5 – Efeito nas recuperações médias do passo de evaporação no processo de
preparação das amostras por SBSE(P20)‐LD seguido de HPLC‐DAD. 185 Figura 10.6 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para as hormonas em estudo na optimização da velocidade de agitação. 186 Figura 10.7 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para as hormonas em estudo na optimização do tempo de extracção. 187 Figura 10.8 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para as hormonas em estudo na optimização do pH da matriz. 187 Figura 10.9 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para as hormonas em estudo na optimização da força iónica da matriz aquosa. 188 Figura 10.10 – Recuperações médias obtidas pela metodologia SBSE(P20)‐LD/HPLC‐DAD para as hormonas em estudo na optimização do conteúdo orgânico da matriz aquosa. 189
Figura 10.11 – Recuperações médias obtidas para as hormonas em estudo sob condições
experimentais optimizadas de acordo com a metodologia SBSE(P20)‐LD/HPLC‐DAD desenvolvida.
190
Figura 10.12 – Comparação das recuperações médias normalizadas ao volume polimérico
envolvido obtidas para as oito hormonas esteróides usando SBSE(PDMS, 126 µL) e SBSE(P20, 86 µL), seguido de LD‐HPLC‐DAD, sob condições experimentais optimizadas para cada fase polimérica.
191
Figura 11.1 – Abundâncias médias obtidas pela metodologia HSSE(P20)‐LD/LVI‐GC‐MS na
optimização do solvente de LD. 198
Figura 11.2 – Abundâncias médias obtidas pela metodologia HSSE(P20)‐LD/LVI‐GC‐MS na
optimização do tempo do tratamento US no passo de LD. 199
xxiii
Figura 11.3 – Abundâncias médias obtidas pela metodologia HSSE(P20)‐LD/LVI‐GC‐MS na
optimização da temperatura de extracção. 200
Figura 11.4 – Abundâncias médias obtidas pela metodologia HSSE(P20)‐LD/LVI‐GC‐MS na
optimização do tempo de extracção. 201
Figura 11.5 – Imagens exemplificativas de frascos de amostragem contendo amostras de café
utilizados no processo extractivo por HSSE(P20) (a) e HSSE(PMDS) (b). 202
Figura 11.6 – Esquema relativo às técnicas de extracção aplicadas a amostras de café no modo
de HS; HS‐SPME(CAR/PDMS, 75 µm) (a); HSSE(P20, 32 µL) (b); HSSE(PDMS, 126 µL) (c). 202
Figura 11.7 – TIC obtidos após extracção por HSSE(P20)‐LD (a) e HSSE(PDMS)‐LD (b) seguida de
análise por LVI‐GC‐MS, sob condições experimentais optimizadas. 203
Figura 11.8 – Abundâncias médias obtidas para as várias famílias químicas usando diferentes
técnicas de extracção. 203
Figura A1.1 – Curvas de calibração instrumental obtidas por HPLC‐DAD para os sete herbicidas
triazínicos estudados. III
Figura A1.2 – Curvas de calibração experimental obtidas por SBSE(PU)‐LD/HPLC‐DAD para os
sete herbicidas triazínicos estudados. IV
Figura A1.3 – Curvas de calibração obtidas por SBSE(PU)‐LD/HPLC‐DAD para análise dos sete
herbicidas triazínicos na amostra de água superficial. V
Figura A1.4 – Curvas de calibração obtidas por SBSE(PU)‐LD/HPLC‐DAD para análise dos sete
herbicidas triazínicos na amostra de água subterrânea. VI
Figura A2.1 – Curvas de calibração instrumental obtidas por HPLC‐DAD para os cinco
metabolitos triazínicos estudados. IX
Figura A2.2 – Curvas de calibração experimental obtidas por SBSE(PU)‐LD/HPLC‐DAD para os
cinco metabolitos triazínicos estudados. X
Figura A2.3 – Curvas de calibração obtidas por SBSE(PU)‐LD/HPLC‐DAD para análise dos cinco
metabolitos triazínicos na amostra de água de consumo. XI
Figura A2.4 – Curvas de calibração obtidas por SBSE(PU)‐LD/HPLC‐DAD para análise dos cinco
metabolitos triazínicos na amostra de água superficial. XII
Figura A2.5 – Curvas de calibração obtidas por SBSE(PU)‐LD/HPLC‐DAD para análise dos cinco
metabolitos triazínicos na amostra de água subterrânea. XIII
xxiv
Índice de Tabelas
Tabela 6.1 – Condições de HPLC‐DAD utilizadas para análise de simazina, atrazina e
terbutilazina, durante a optimização da formulação da espuma de PU. 92 Tabela 6.2 – Condições de HPLC‐DAD utilizadas para análise de herbicidas triazínicos. 92 Tabela 6.3 – Condições de HPLC‐DAD utilizadas para análise de metabolitos triazínicos. 93 Tabela 6.4 – Condições de HPLC‐DAD utilizadas para análise de hormonas esteróides 93 Tabela 7.1 – Formulação base (P1) da espuma de PU. 103 Tabela 7.2 – Resultados obtidos para a massa específica aparente e recuperação média de ATZ para os polímeros obtidos por variação das proporções de água e DC193. 103 Tabela 7.3 – Resultados obtidos para a massa específica aparente e recuperação média de ATZ para os polímeros obtidos por introdução de glicerol na formulação. 105 Tabela 7.4 – Resultados obtidos para a massa específica aparente e recuperação média de ATZ para os polímeros obtidos por introdução de glicerol na formulação, com variação simultânea da quantidade de água. 106 Tabela 7.5 – Resultados obtidos para a massa específica aparente e recuperação média de ATZ para os polímeros obtidos por introdução de EDA‐PO‐EO na formulação. 106 Tabela 7.6 – Resultados obtidos para a massa específica aparente e recuperação média de ATZ para os polímeros obtidos por introdução de TMPE na formulação. 107 Tabela 7.7 – Resultados obtidos para a massa específica aparente e recuperação média de ATZ para P1 e P23. 108 Tabela 7.8 – Formulações e resultados obtidos para as recuperações médias obtidas por cada polímero em meio aquoso para a mistura de SMZ, ATZ e TBZ (SBSE: 18h a 1000 rpm; LD: 5 mL de MeOH, 20 min de tratamento US). 109 Tabela 7.9 – Valores de % de MDI, massa específica aparente e recuperação média para a ATZ obtidos para os polímeros sintetizados apenas com glicerol e PPG (10 g de PPG). 110
Tabela 7.10 – Valores das densidades (kg/dm3) e dos volumes médios (µL) obtidos por
picnometria para os polímeros seleccionados. 113
xxv
Tabela 7.11 – Valores calculados para o volume aberto, % de células abertas e ligações ureia de
P20. 113
Tabela 7.12 – Formulações dos polímeros sintetizados com diferente óleo de silicone. 123
Tabela 7.13 – Valores obtidos para a densidade dos polímeros e valores calculados para o
volume aberto, % de células abertas, ligações ureia, volume médio de uma barra de PU e recuperação média de ATZ para P20, P24, P25 e P26.
123
Tabela 7.14 – Adsorção de furano em P20. 132
Tabela 8.1 – Resumo dos valores de log KO/Westimados para as triazinas em estudo. 137
Tabela 8.2 – Resumo das gamas de trabalho, coeficientes de correlação (r2), LODs, LOQs e precisão instrumental (RSD) obtidos para cada composto por HPLC‐DAD, sob condições instrumentais optimizadas.
140
Tabela 8.3 – Condições de SBSE(LD)‐PU optimizadas para extracção de herbicidas triazínicos. 147
Tabela 8.4 – Resumo das gamas de trabalho, coeficientes de correlação (r2), LODs, LOQs e valores de precisão (RSD) obtidos por SBSE(P20)‐LD/HPLC‐DAD, sob condições experimentais optimizadas.
148
Tabela 8.5 – Coeficientes de distribuição determinados para P20 e PDMS para os herbicidas
triazínicos em estudo. 152
Tabela 8.6 – Declives, coeficientes de correlação (r2) e valores obtidos nas amostras para os sete herbicidas triazínicos em estudo, usando a metodologia optimizada SBSE(P20)‐LD/HPLC‐DAD. 153
Tabela 9.1 – Resumo dos valores de log KO/We pKapara os metabolitos triazínicos em estudo. 160
Tabela 9.2 – Resumo das gamas de trabalho, coeficientes de correlação (r2), LODs, LOQs e precisão instrumental (RSD) obtidos por HPLC‐DAD, sob condições instrumentais optimizadas. 163
Tabela 9.3 – Condições de SBSE(LD)‐PU utilizadas para extracção de metabolitos triazínicos. 171
Tabela 9.4 – Resumo das gamas de trabalho, coeficientes de correlação (r2), LODs, LOQs e valores de precisão instrumental (RSD) obtidos por SBSE(P20)‐LD/HPLC‐DAD, sob condições experimentais optimizadas.
173
Tabela 9.5 – Coeficientes de distribuição determinados para P20 e PDMS para os metabolitos
triazínicos em estudo. 174
Tabela 9.6 – Declives, coeficientes de correlação (r2) e valores obtidos nas amostras para os cinco metabolitos triazínicos em estudo, usando a metodologia optimizada SBSE(P20)‐LD/HPLC‐ DAD.
175
Tabela 10.1 – Resumo dos valores de log KO/Wpara as hormonas em estudo.3 181
xxvi
Tabela 10.3 – Coeficientes de distribuição determinados para P20 e PDMS para as hormonas
esteróides em estudo. 191
Tabela 11.1 – Composições médias relativas observadas para os compostos aromáticos de
amostras de café, obtidas por HS‐SPME(CAR/PDMS), HSSE(P20) e HSSE(PDMS), com análise por LVI‐GC‐MS.
204
xxvii
Abreviaturas
α – Selectividade ou factor de separação β – Razão de fase µL – Microlitros λ – Comprimento de onda Aaquosa – Analito A na fase aquosa Aorgânica – Analito A na fase orgânica Apadrão – Área do analito no padrão de fortificação Aapós extracção – Área do analito após extracção ACN – Acetonitrilo ATR – Reflectância total atenuada (attenuated total reflectance)ATZ – Atrazina C8 – Grupo n‐octilo C18 – Grupo n‐octadecilo C0 – Concentração do analito na amostra real CE – Concentração do analito na fase estacionária Cf – Concentração do analito na fibra de SPME Ci – Concentração inicial do analito CM – Concentração do analito na fase móvel CPDMS – Concentração do analito no PDMS Cs – Concentração do analito na amostra CW – Concentração do analito na fase aquosa CAR – Carboxen Da – Dalton DAD – Rede de díodos DBTL – Dilaurato de dibutilestanho (dibutyltin dilaurate) DC – Voltagem constante DDA – Didealquiltrazina DEA – Desetilatrazina DIA – Desisopropilatrazina DTBZ – Desetilterbutilazina DVB – Divinilbenzeno ECD – Detector de captura electrónica (electron capture detector) EDA‐PO‐EO – Etilenodiamina tetrakis (propoxilato‐block‐etoxilato) tetrol
xxviii
EDC – Composto desregulador endócrino (endocrine disrupting chemical) EI – Ionização electrónica (electronic ionization)
ESI – electropulverização (electrospray)
EPA – Agência de Protecção Ambiental Norte‐Americana (United States Environmental
Protection Agency)
EU – União Europeia (European Union) EUA – Estados Unidos da América
FID – Detector de ionização de chama (flame ionization detector)
FTIR – Espectroscopia de infravermelho com transformada de Fourier (Fourier transform
infrared‐espectroscopy)
GC – Cromatografia gasosa (gas chromatography)
H – Altura do prato teórico
HPLC – Cromatografia líquida de alta eficiência (High Performance Liquid Chromatography) HS‐SPME – Microextracção em fase sólida no espaço de cabeça (headspace solid phase
microextraction)
HSSE – Extracção sortiva no espaço de cabeça (headspace sorptive extraction)
IUPAC – União Internacional de Química Pura e Aplicada (International Union of Pure and
Applied Chemistry) IR – Infravermelho (infrared) K – Coeficiente de distribuição k’ – Factor de capacidade Ka – Constante de acidez Kfs – Coeficiente de distribuição entre a fibra de SPME e amostra KPDMS/W – Coeficiente de distribuição dos analitos entre o PDMS e a fase aquosa KO/W – Coeficiente de distribuição octanol‐água KPU/W ‐ Coeficiente de distribuição dos analitos entre o PU e a fase aquosa LC – Cromatograma líquida (liquid chromatography) LD – Dessorção líquida (liquid desorption) LLE – Extracção líquido‐líquido (liquid‐liquid extraction) LOD – Limite de detecção (limit of detection) LOQ – Limite de quantificação (limit of quantification) LVI – Injecção de grande volume (large volume injection) mPDMS – Massa de analito no PDMS mW – Massa de analito na fase aquosa m0 – Quantidade total de analito inicialmente presente na amostra aquosa
xxix m/v – Massa por volume m/z – Razão massa/carga MDI – 4,4’‐Difenilmetano diisocianato MeOH – Metanol MM – Massa molecular MS – Espectrometria de massa (mass spectrometry) n – Número de moles N – Eficiência da coluna cromatográfica NCO/OH – Índice de isocianato NPD – Detector de azoto e fósforo (nitrogen phosphorus detector) OCDE – Organização para a Cooperação e Desenvolvimento Económico ODS – Octadecilsílica OH‐ATR – 2‐Hidroxiatrazina OH‐DEA – Desetil‐2‐hidroxiatrazina OH‐TBZ – 2‐Hidroxiterbutilazina OMS – Organização Mundial de Saúde oop – Fora do plano (out of plane) PAH – Hidrocarboneto aromático policíclico (polycyclic aromatic hydrocarbon) PDMS – Polidimetilsiloxano ppb – Partes por bilião PPG – Propoxilato de glicerol ppt – Partes por trilião PTFE – Politetrafluoretileno
PTV – Injector de vaporização com temperatura programada (programmed temperature
vaporization) PU – Poliuretano r2 – Coeficiente de correlação R – Grupo substituinte RS – Resolução Recup. – Recuperação RF – Voltagem de radiofrequência rpm – Rotações por minuto RSD – Desvio padrão relativo (relative standard deviation) S/SL – Split/splitless S/N – Razão sinal/ ruído
xxx
SAM – Método de adição de padrão (standard addition method)
SBSE – Extracção sortiva em barra de agitação (stir bar sorptive extraction)
SDBS – Base de dados espectrais para compostos orgânicos (spectral database for organic
compounds) SEM – Microscopia electrónica de varrimento (scanning electron microscopy) SIM – Monitorização seleccionada de iões (selected ion monitorization) SMZ – Simazina SPE – Extracção em fase sólida (solid‐phase extraction) SPME – Microextracção em fase sólida (solid‐phase microextration) st – Stretching tM – Tempo morto tR – Tempo de retenção tR’ – Tempo de retenção ajustado TBZ – Terbutilazina TCD – Detector de condutividade térmica (TCD) TD – Dessorção térmica TDI – Diisocianato de tolueno TIC – Traçados de corrente iónica TMPE – Trimetilolpropano etoxilato u – Velocidade linear US – Ultrassónico UV‐vis – Ultravioleta‐visível Va – Volume ou conteúdo em células abertas Ve – Volume externo Vf – Volume da fibra de SPME Vg – Volume determinado com gás ou volume de espuma não acessível ao gás VPDMS – Volume de PDMS Vs – Volume da amostra VW – Volume de amostra v/v – Volume por volume VMA – Valor máximo admissível ܹ – Largura da banda ܹଵ ଶ ൗ – Largura da banda a meia altura WCOT – Colunas tubulares de parede revestida (Wall‐Coated Open Tubular Columns)
1
Técnicas de Extracção Sortiva
3
1.1. Introdução Geral
A preparação de amostra é um processo cujo principal objectivo consiste em transferir os compostos para uma forma mais adequada à sua detecção. Este processo pode ou não envolver a extracção do composto de uma matriz complexa para um meio mais apropriado à sua análise, concentração para ganho de sensibilidade, remoção de espécies interferentes ou mesmo derivatização de um composto. Nas últimas décadas têm ocorrido diversas inovações nas técnicas de preparação de amostra com o intuito de diminuir o tempo de análise e facilitar a manipulação laboratorial. Para além disso torna‐se prioritário reduzir o gasto das quantidades de solventes tóxicos envolvidos neste tipo de técnicas, devido ao impacto ambiental que isso acarreta, no âmbito dos actuais conceitos da designada “química verde”.1‐4
Muitas das metodologias implementadas já não se coadunam com as exigências de redução de tempo analítico, com vista a uma maior automatização e maior eficácia do trabalho na rotina laboratorial. Existe uma tendência para a miniaturização analítica, que se tem verificado na redução da quantidade de solventes utilizados, assim como do volume de amostra, facilidade de automatização e consequente ganho em tempo analítico. Neste contexto, destacam‐se as técnicas de extracção em fase sólida (SPE), microextracção em fase sólida (SPME) e mais recentemente, a extracção sortiva em barra de agitação (SBSE). Para além de ambientalmente favoráveis, estas técnicas permitem uma redução da manipulação analítica, elevadas sensibilidade e reprodutibilidade, rapidez, baixo custo e facilidade de automatização.3
1.2. Extracção em Fase Sólida (SPE)
As técnicas de SPE foram introduzidas nos anos 70 do século XX, quando surgiram no mercado pequenas colunas, normalmente designadas por cartuchos, empacotadas com materiais sólidos. De um modo geral, faz‐se passar a amostra líquida contendo os compostos com interesse através de uma fase sólida que os retém selectivamente, sendo posteriormente recolhidos com recurso à passagem de um solvente com polaridade adequada. Os princípios são semelhantes aos da cromatografia líquida (HPLC), sendo os compostos separados de acordo com os seus coeficientes de partição entre as fases móvel e estacionária. Os estudos realizados com SPE revelaram que esta técnica permite analisar uma grande variedade de