New developments in pathogen detection and identification
Texto
Imagem




Documentos relacionados
The universal primers designed by Folmer and colleagues [1] (HCO2198- LCO1490, henceforth named “ Folmer primers ” ) for amplification of a 658 base pair (bp) fragment of the 5 ′ end
28,29 described a rapid and simple method for the identification of HIV-1 subtypes by PCR amplification of a region of gp41 using subtype-specific primers.. Therefore, our aim was
PCR amplification using specific primers amplified a sin- gle 2.5 kb fragment in Brassica lines and the DNA se- quences of the fragments showed similarity to NBS-LRR domains present
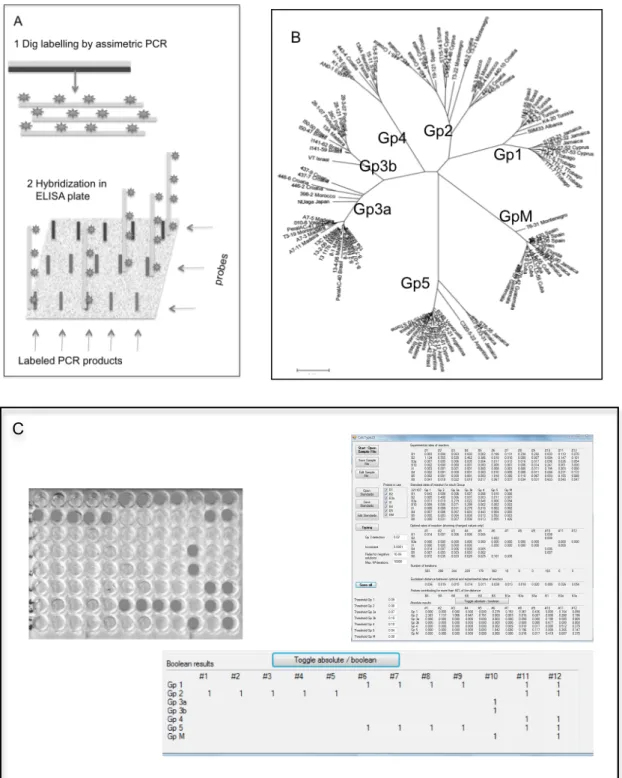
In this study, we used IS 6110 as the specific identification target to develop a novel hybridization signal amplification method (HSAM) for the rapid and
In this study, the development of a methodology based on nested-PCR technique, using an external primers pair in a first amplification and the “classical” primers pair MY09/11 in
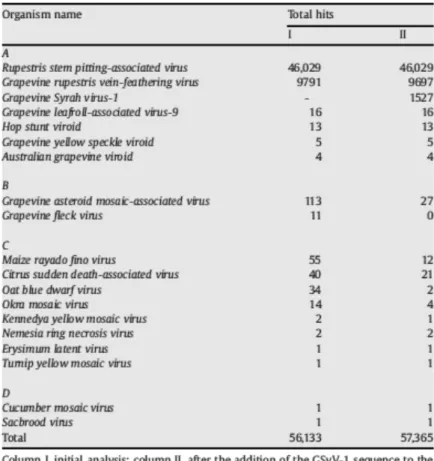
A rolling-circle amplification protocol tested in this study allowed specific detection and identification of an episomal BSV isolate infecting Nanicão Jangada cultivar,
The selected primers (Table 1) generated a total of 161 scorable amplification products that were analyzed and used as variables in our analysis.. The variation between Crotalus and
The aim of this study was to develop and evaluate a padlock probe based on the Rolling Circle Amplification (RCA), which targeted to 16S-23S rDNA region of S. The specificity
