Rapid MALDI-TOF mass spectrometry strain typing during a large outbreak of Shiga-Toxigenic Escherichia coli.
Texto
Imagem

Documentos relacionados
Growth (A) and ethanol production (B) by Escherichia coli strain KO11 (closed symbols) and Klebsiella oxytoca strain P2 (opened symbols) during sugarcane juice fermentation...
(a) Number of species (%); (b) mean species richness (± SE); (c) abundance (%) and (d) mean abundance (± SE) of Testate amoebae, Rotifera, Cladocera and Copepoda during low,
Characteristics of simulated grazing sample by cattle on signal grass managed at a mean height of 20 cm, in continuous stocking, during the rainy period.. Sample of simulation
Values expressed as mean ± standard deviation of concentric peak torque (Panel A), Eccentric peak torque (Panel B), Changes (%) of concentric (Panel C) and eccentric (Panel D)
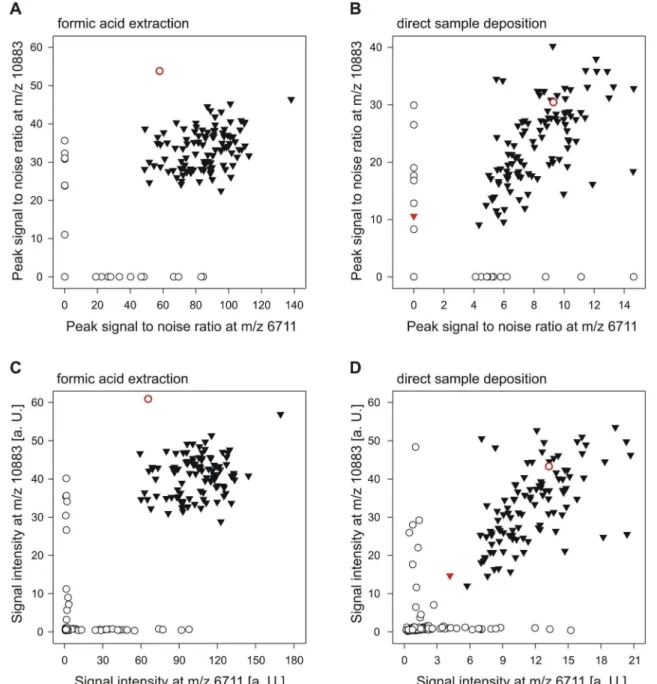
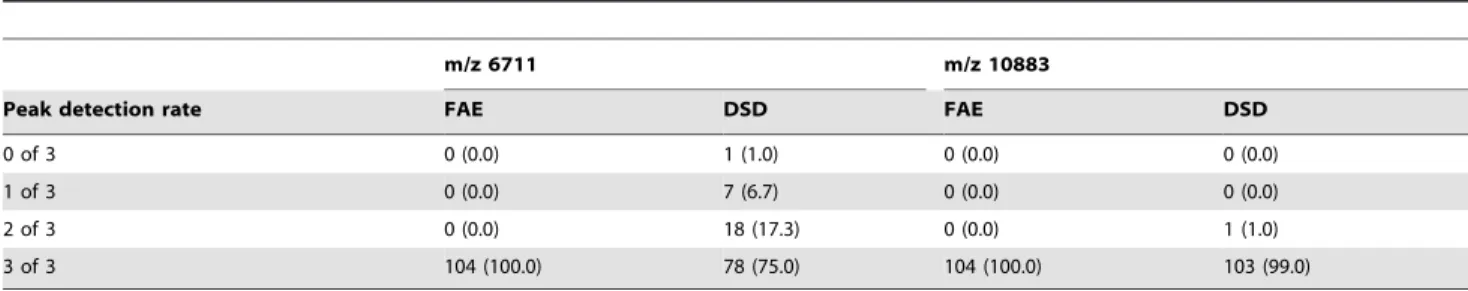
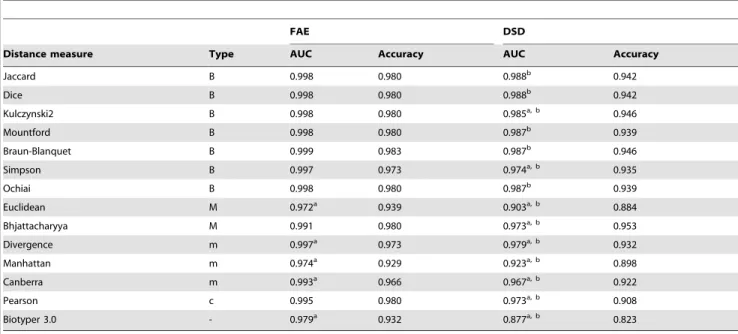
In this pilot study, using a simple preparation method and the algorithm for sample classification implemented in the MALDI-ToF analysis software, we obtained acceptable
Spatio-temporal distributions of the wind speed (a), wind direction angle (b), vertical component of the wind vector (c) and signal-to-noise ratio (d) obtained from measurements of
at 47 mbar with two proportional valves in a feedback loop configuration up- and down- stream of the optical cavity at a temperature of 80 ◦ C. The high signal to noise ratio
coli collected during an outbreak at the Uni- versity hospital Basel, Switzerland, [ 10 ] by performing MALDI-TOF MS based typing and comparing the results to those obtained
