w w w.jo u r n als . e l s e vie r . c o m / r e v i s t a - b r a s i l e i r a - d e - f a r m a c o g n o s i a
Original
Article
Penicillosides
A
and
B:
new
cerebrosides
from
the
marine-derived
fungus
Penicillium
species
Samar
S.A.
Murshid
a,
Jihan
M.
Badr
a,b,
Diaa
T.A.
Youssef
a,∗aDepartmentofNaturalProducts,FacultyofPharmacy,KingAbdulazizUniversity,Jeddah,SaudiArabia
bDepartmentofPharmacognosy,FacultyofPharmacy,SuezCanalUniversity,Ismailia,Egypt
a
r
t
i
c
l
e
i
n
f
o
Articlehistory: Received5May2015 Accepted17September2015 Availableonline10November2015
Keywords: Didemnum Penicillum Cerebrosides Penicillosides Antimicrobialactivity HeLacells
a
b
s
t
r
a
c
t
Inthecourseofourongoingefforttoidentifybioactivecompoundsfrommarine-derivedfungi,the marinefungus,PenicilliumspecieswasisolatedfromtheRedSeatunicate,Didemnumspecies.Twonew cerebrosides,penicillosidesAandBwereisolatedfromthemarine-derivedfungus,Penicilliumspecies usingdifferentchromatographicmethods.Theirstructureswereestablishedbydifferentspectroscopic dataincluding1D(1HNMRand13CNMR)and2DNMR(COSY,HSQC,andHMBC)studiesaswellas
high-resolutionmassspectraldata.PenicillosideAdisplayedantifungalactivityagainstCandidaalbicanswhile penicillosideBillustratedantibacterialactivitiesagainstStaphylococcusaureusandEscherichiacoliinthe agardiffusionassay.Additionally,bothcompoundsshowedweakactivityagainstHeLacells.
©2015SociedadeBrasileiradeFarmacognosia.PublishedbyElsevierEditoraLtda.Allrightsreserved.
Introduction
Marinemicroorganismshavereceivedagreatattentionlately; accordingly,thefungibegan toberecognizedasaliable source ofpotentially usefulnaturalproducts(Fenical,1993; Bugniand Ireland,2004).Althoughstudiesontheseorganismsbeganmuch later than theircounterparts in terrestrial environments, more thanahundrednovelcompoundshavebeenfoundannuallysince the late 1990s (Blunt et al., 2013). The high number of com-poundsreportedfromthegenusPenicilliumcouldbejustifiedby thefactthatitsdifferentspeciesaresalttolerant,fastgrowingand areobtainedeasilyfrommanysubstrates.Thispromptedmany researchers to investigate variable Penicillium species isolated fromdifferent habitats.Their extensivestudiesconcerned with biological activitiesof the isolated secondary metaboliteswere extremelyefficient.Amongthesignificantactivitiesreportedwere theantibacterial(Qietal.,2009;Devietal.,2012;Abo-Kadoum etal.,2013;Subramanietal.,2013),cytotoxicandanticancer(Wang etal.,2009a,b;Sunetal.,2012;Gaoetal.,2013;Abo-Kadoumetal., 2013;Subramanietal.,2013).
Inthecourseofourongoingsearchforbioactivecompounds fromRedSeamarine-derivedfungi,thefungusPenicilliumspecies wasisolatedfromthetunicateDidemnumspeciesandwascultured
∗ Correspondingauthor.
E-mail:[email protected](D.T.A.Youssef).
inSabourauddextrosebroth.Fungalmyceliawereextractedand fractionatedusingdifferentchromatographictechniquestoafford twocompounds.Basedondifferentspectroscopicdataincluding HRESIMS,1D(1HNMRand13CNMR)and2DNMR(COSY,HSQC, andHMBC),thestructuresofthecompoundswereestablishedas cerebrosidesand namedpenicillosides A(1)and B(2).The iso-latedcompoundswereevaluatedfortheirantimicrobialactivities against different pathogens and theircytotoxic activityagainst HeLacells.PenicillosidesAandBdisplayedsignificant antimicro-bialactivitiesagainstCandidaalbicans;Staphylococcusaureusand Escherichiacolirespectivelyintheagardiffusionassay.Additionally, bothcompoundsshowedweakactivityagainstHeLacells.
Materialsandmethods
Generalexperimentalprocedures
OpticalrotationwasmeasuredonaJASCOdigitalPolarimeter. 1Dand2DNMRspectra(chemicalshiftsinppm,couplingconstants inHz)wererecordedonBrukerAvanceDRX600MHz spectrome-tersusingCD3ODassolvents.NormalandHRESIMSspectrawere recordedonaLTQOrbitrapandanAPI2000(ThermoFinnigan, Bre-men,Germany)massspectrometers.Forcolumnchromatography, silicagel(Merck,70–230meshASTM)andSephadexLH-20 (Phar-macia)wereused.Pre-coatedsilicagel60 F-254plates(Merck) were used for TLC. The HPLC separation was performed on a RP18, 250mm×10mm, 5m Phenomenex Luna column using
http://dx.doi.org/10.1016/j.bjp.2015.09.007
CH3CN/H2Ogradientasmobilephaseat220nmandataflowrate of2.0ml/min.
Collectionofthehosttunicateandpreparationofthefungalisolate
Themarinetunicate Didemnumspecies wascollectedin the Mangrovelocatedin Sharm El-SheikhontheEgyptian RedSea coastatdepthof1–2mduringJuly2010.Inordertoensure fun-galisolatestobeendophyticwhenobtained,asurfacesterilization oftunicatewasperformed.Thetunicatesamplewasdisinfected with5% sodiumhypochlorite, followed by70% ethanol (Li and Wang,2009),toensurethatepiphyticfungiweredestroyedbythe washingwhileassociatedfungi(ifany)werenotaffected. Approxi-mately2cm3ofinnertissueoftunicatematerialwashomogenized usingasterilemortarandpestlecontaining10mlofsterile artifi-cialseawaterunderasepticconditions.Theresultinghomogenate wasdilutedwithsterileseawateratthreedilutions(1:10,1:100, and 1:1000). For fungicultivation, 100l of each dilution was platedin quadruplicate onto fourplates of each of the follow-ingmedia;Czapek-Doxyeastagarmedium(NaNO33g,KCl0.5g, K2HPO4 0.1g,MgSO4·7H2O0.5g,FeSO4 0.01g,sucrose30g,agar 20g,pH6.7);maltagarmedium(maltextract17g,peptone3g, agar20g)andSabourauddextroseagarmedium.Allmediawere amendedwith2%NaCland0.25%chloramphenicolasantibacterial agenttopreventbacterialgrowthandtoenrichfungigrowth.Plates werewrappedinparafilm,incubatedat28◦Cfor1–3weeksuntil
themorphologyoffungicouldbedistinguished.Manypurification stepsweredoneuntilpurefungalisolateswereobtained.
Identificationoffungalstrain
ExtractionofgenomeDNAfromculturedfungalisolate
Thefungalisolatewasculturedincorrespondingbrothat28◦C
for 2–5 days. The mycelia were harvested separately by using vacuumfiltrationanddriedwithtwolayersofpapertowel.The resultingmycelialmatwasgroundintopowderwithliquid nitro-gen.ThefungalDNAwasextractedusingQIAampDNAMiniKit (Qiagen)accordingtomanufacturer’sinstructions.
AmplificationoffungalITS-rDNAfragmentsofisolate
The genomic DNA of the fungal strain was used as the template to amplify fungal ITS-rDNA fragments using the primers ITS1 (5′-TCCGTAGGTGAACCTGCG-3′) and ITS4 (5′
-TCCTCCGCTTATTGATATGC-3′) (White et al., 1990) which were
synthesizedbytheUniversityofUtahDNA/peptidesynthesiscore facility.ThereactionmixtureforPCRamplificationcontained5lof 10×reactionbufferwith15mMMgCl2(Invitrogen),2lof2.5mM dNTPs,0.5lof10Meachprimer,4loffungalDNA,0.3lof TaqDNApolymerase(5Ul−1,Invitrogen),and39.7lofH2O.PCR conditionsincludedaninitialdenaturationat94◦Cfor4min
fol-lowedby30cyclesofdenaturationat94◦Cfor50s,annealingat
51◦Cfor50s,andelongationat68◦Cfor1min,withafinal
elon-gationat68◦Cfor10min.PCRproductswerepurifiedusingthe
AgaroseGelDNAPurificationKit(Qiagen)andsequencedinatthe UniversityofUtahDNAsequencingfacility.
SequencefungalITS-rDNAregionsofisolate
For preliminaryidentification,sequences offungal ITS-rDNA regionsobtainedfromthemarinetunicateDidemnumspecieswere comparedwithrelatedsequencesinNationalCenterfor Biotech-nologyInformation(NCBI)(http://www.ncbi.nlm.nih.gov).Fungal ITS-rDNAsequencesacquiredinthisstudywereeditedandaligned withthebestn-BLASThitsfromGenBankintheClustalX (ver-sion1.83)program(Thompsonetal.,1997),andfurthermanually adjustedusingBioEditsoftware(Hall,1999).TheprogramMEGA
5(Tamuraetal.,2011)wasappliedtocalculatethebase composi-tionofthefungalsequences.Thesequenceanalysisbasedon99% sequenceidentitywithPenicilliumspeciesRsf-2(NCBIaccession numberEF660439.1).
Isolationandpurificationofcompounds1and2
Large scale cultureof the marine-derived fungusPenicillium specieswascarriedout.Theculturewasincubatedatroom tem-peraturefor30days.Afterthat,250mlofEtOAcwereaddedto eachflaskleftovernighttostopcellgrowth.Culturemediaand myceliawereseparatedbyvacuumfiltrationusingBuchnerfunnel. ThemyceliawereleftinMeOHovernightforextraction.Theextract wasdriedunderreducedpressure.Theviscousextract(1200mg) wasdissolvedin250mlof70%MeOHthenextractedwith hex-ane(3ml×100ml).Themethanollayerwasconcentratedunder reducedpressuretoyieldanotherviscousbrownresidue(765mg). Aportionofthealcoholicextract(600mg)wasfractionatedover silicagel(VaccumLiquidChromatography)usingCHCl3/MeOH gra-dient(100%CHCl3then2,5,7,10,15%MeOHinCHCl3)togivesix mainfractions.Fraction2(65mg,elutedwith2%MeOHinCHCl3) waspurifiedonSephadexLH-20,elutedwithMeOHtoobtainfinally tensub-fractions.Finalpurificationofthemainsubfractionswas achievedbyHPLCusinggradientsystemstartedwith5%ACN/H2O to100%ACNin15mintoaffordcompound1(3mg).Fraction3 (145mg)wasdissolvedinMeOHandsubjectedtoSephadex LH-20columnelutedwith100%MeOH.Thefractionsshowingdistinct spotswerepurifiedonsilicagelusingpet.ether/CHCl3/MeOH gra-dient(100%pet.Ether,followedby20,40,60,80%CHCl3 inpet. Ether,followedby5,10,15,20%MeOHinCHCl3).Fractionseluted with5%MeOHinCHCl3offeredimpure2.Finalpurificationwas performedbyHPLCusinggradientsystemfrom10%ACN/H2Oto 100%ACNover20mintoobtaincompound2(23.1mg).
Biologicalevaluationofcompounds1and2
Determinationoftheantimicrobialactivityofcompounds1and2 TheprocedurewasconductedintriplicateaccordingtoValgas etal.(2007)andSinghandJain(2011).Compounds1and2were testedforantibacterialactivityagainstaGrampositivebacterium (StaphylococcusaureusATCC25923),aGramnegativebacterium (Escherichiacoli ATCC25922), and yeast(CandidaalbicansATCC 14053)usingagardiffusionmethod.Accuratelymeasured0.1ml (100gdissolvedinDMSO)ofeachcompoundwereinsertedin thecups then incubatedat 37◦C for24h. Theinhibition zones
weremeasuredandcomparedwiththereferenceantibioticsand antifungaldrugs;ampicillin,imipenemandclotrimazole(eachof 10g/discgiving30,30and40mminhibitionzonerespectively).
Determinationofcytotoxicactivityofcompounds1and2
Table1
NMRspectraldataofcompounds1and2(CD3OD).
Position 1 2
ıC(mult.)a ıH(mult.,JinHz) ıC(mult.)a ıH(mult.,JinHz)
1 67.4(CH2) 3.63(m) 69.7(CH2) 4.11,m;3.7,dd(10.2,3.6)
2 54.6(CH) 3.97(m) 54.6(CH) 3.97,m
3 71.5(CH) 3.64(m) 35.9(CH2) 1.69,m;1.5m
4 33.1(CH2) 2.04(m) 26.1(CH2) 1.39,m
5 32.7(CH2) 1.28(m) 40.8(CH2) 1.97,t(7.2)
6 26.5(CH2) 2.78(brt,6.0) 136.8(qC) –
7 131.0(CH) 5.34(brd,14.1) 124.8(CH) 5.14,t(6.6)
8 130.8(CH) 5.32(m) 33.8(CH2) 2.07,m
9 28.2(CH2) 2.05(m) 33.1(CH2) 1.28,m
10 14.5(CH3) 0.89,t(6.6)
11 16.1(CH3) 1.59,s
10–14 29.1–30.8(5×CH2) 1.24–1.33(m)
15 23.7(CH2) 1.24–1.33(m)
16 14.4(CH3) 0.90(t,6.6)
1′ 174.2(qC) – 177.2(qC) –
2′ 72.95(CH) 4.16(m) 72.9(CH) 4.12,d(6.6)
3′ 35.0(CH
2) 2.33(m) 73.1(CH) 3.98,dd(7.8,4.2)
4′ 26.05(CH
2) 1.60(m) 131.1(CH) 5.47,dd(15.6,7.8)
5′ 29.2(CH
2) 2.22(m) 134.6(CH) 5.71,m
6′
130.7(CH) 5.38(m)
7′ 130.9(CH) 5.42(brd,15.0)
8′ 29.1(CH
2) 2.06(m)
9′ 26.5(CH
2) 2.68(m)
10′ 129.0(CH) 5.31(m)
6′-10′ 30.5–33.0(5×CH
2) 1.25–1.35,m
11′ 129.1(CH) 5.29(brd,13.2) 14.4(CH
3) 0.89,t(6.6)
12′
28.1(CH2) 2.05(m)
13′
-16′ 29.1–30.8(4
×CH2) 1.24–1.33(m)
17′ 23.6(CH
2) 1.24–1.33(m)
18′ 14.4(CH
3) 0.90(t,6.6)
1′′ 104.7(CH) 4.26(d,7.2) 104.7(CH) 4.27,d(7.8)
2′′ 75.0(CH) 3.18(m) 75.0(CH) 3.18,m
3′′
77.9(CH) 3.34(m) 77.9(CH) 3.34,m
4′′ 71.8(CH) 3.27(m) 71.5(CH) 3.27,m
5′′ 78.0(CH) 3.28(m) 78.0(CH) 3.28,m
6′′ 62.6(CH
2) 3.86(m),3.67(m) 62.6(CH2) 3.85,dd(12.0,1.2);
3.66,dd(11.4,4.8)
aMultiplicitywasdetectedfromHSQC.
Spectraldata
Penicilloside A (1). Colourless amorphous powder, positive HRESIMS m/z 712.5366 (calculated for C40H74NO9, 712.5364 [M+H]+),[
␣]D−13◦(c1.5,MeOH).NMRdata:seeTable1. Penicilloside B (2). Colourless amorphous powder; positive HRESIMS m/z 546.3646 (calculated for C28H52NO9, 546.3642 [M+H]+),[
␣]D−19.6◦(c1.5,MeOH).NMRdata:seeTable1.
Resultsanddiscussion
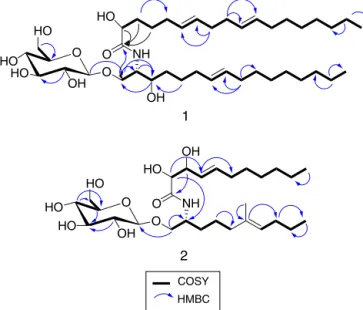
Compound 1 was isolated as a colourless amorphous pow-der. Its molecular formula was suggested as C40H73NO9 based on different spectral data including HRESIMS (m/z 712.5366, [M+H]+), 1Dand2DNMRdata(Table1).Differentspectraldata declaredthepresencefor asugarmoiety,anamidelinkageand aliphatic chains, thus supporting its cerebroside nature (Wang etal.,2009a,b).Analysisof1HNMRand13CNMRdataprovedthe presenceofsignalsofglucosemoiety,where1H NMRspectrum revealedsignalsresonatingintherangeof3.18–4.26ppm.HSQC correlatedtheseprotonstotheircorrespondingcarbonsdetected in the range of 62.6–104.7ppm. The coupling constant of the anomericproton(J=7.2Hz)confirmedtheˇ-configurationofthe glucose moiety (Renet al.,2009; Sheet al., 2009; Peng et al., 2011).Carefulexaminationofdifferentspectraldataallowedthe assignmentoftwosubunits;subunitA(C-1→C-16)andsubunit B(C-1′
→C18′).FromtheCOSYandHSQC experiments,subunit
Awasassignedas onemethyl,oneoxygenatedmethylene, two
downfield shifted methines, two protonated sp2 carbons and ten methylenes. From 2D NMR spectra, C-3 wasproved to be oxygenatedasestablishedfromthechemical shiftatıC 71.5/ıH 3.64.Theothermethinewasconfirmedtobelinkedtotheamide group,thehydroxylatedmethine(C-3)andtheoxygenated methy-lene(ıC67.4/ıH3.63).Thus,thefragementfromC-1toC-3was proved. Thelocationof thesp2 carbonswassuggestedtobeat C-7/C-8separatedfromC-3bythreemethylenes.Thissuggestion was confirmed from spin–spin coupling between H-7 (ı 5.34) andH2-6(ı2.78)whichinturnwascoupledwithH2-5(ı1.28). AlsoH2-4 (ı 2.04)wascoupledwithH-3(ı 3.64).Additionally, HMBCrevealedcross peaksfromH2-9(ı2.05)toC-8(ı130.8) andtoC-7(ı131.0),confirmingtheassignmentofthefragment C-1toC-8.Thelastpartofsubunit(A)wasfoundtoconsistofa terminalmethyl(ıC14.4/ıH0.90)whichwascoupledtoC-15;the adjacent methylene. H-8 revealed spin–spincoupling with C-9 (theadjacentmethylene).Thenumberofthemethylenegroups betweenC-8andtheterminalmethylC-16wasfoundtobeseven asindicated fromMSspectrum, throughthefragmentdetected at m/z 599 corresponding to a loss of the terminal methyl in additiontosevenmethylenes.Accordingly,subunitA(C-1toC-16) structurewasconfirmed.ThechemicalshiftvaluesofH2-1,H-2, H-3together withtheircorresponding carbonsare comparable tothosepreviouslyreportedforsimilarcerebrosides(Pengetal., 2011).SubunitBwasprovedtoconsistofeighteencarbons(C-1′
toC-18′).HMBCdeclaredcrosspeaksfromeachofH-2′(ı4.16),
HO
HO
OH
OH
OH
HO HO
O O
O OH
2
1
COSY
NH NH
HMBC
HO
HO HO
HO
O O
O
Fig.1.SelectedCOSYandHMBCcorrelationsofcompounds1and2.
protonatedsp2carbonsweredetectedinfragmentB.Their loca-tionwasdefinedatC-6′/C-7′andC-10′/C-11′.Thenoninterrupted
spin–spincoupling from H2-3′ to H-6′ and from H-7′ to H-10′ supportedthis assumption. A furtherconfirmation was gained fromHMBCwhichrevealedcrosspeaksfromH2-8′(ı2.06)toeach ofC-6′(ı130.7)andC-7′(ı130.9)andfromH
2-9′(ı2.68)toeach ofC-10′(ı129.0)andC-11′(ı129.1).Theterminalmethylgroup
(CH3-18)waslinkedtoH2-17′ andC-17′ asdeclaredfromCOSY andHMBCrespectively(Fig.1).The1Hand13CNMRdataforC-1′,
C-2′andH-2′aretypicalforthosepreviouslyreportedforsimilar
cerebrosides(Elkhayatetal.,2012).TheconnectionofsubunitA withBwasconfirmedfromHMBCthatshowedcrosspeaksfrom H-3toC-1′.AdditionalcrosspeakfromH-1′′toC-1confirmedthe
attachmentofsubunitAwiththeglucosemoiety.Basedonthe pre-viousdiscussion,thestructureofcompound1wasestablished.This isthefirstreportofthiscompoundfromnaturalsource.Therefore, itisconsideredasanewnaturalproductandnamedpenicillosideA.
HO
HO HO
HO
O O
O OH
OH
OH
OH HO
HO HO
HO
O O
O NH
2
1
NH
Compound2wasisolatedasacolourlessamorphouspowder. ItsmolecularformulawassuggestedasC28H51O9Nbasedon dif-ferentspectraldataincludingHRESIMS(m/z546.3646[M+H]+),1H NMR,13CNMRandHSQC(Table1).Thecerebrosidenatureof com-pound2wassuggested from1DNMR datawhich declaredthe presenceofasugarresidue,oneormorealiphaticchainsandan amidelinkage(Elkhayatetal.,2012).Analysisof1H–1HCOSY spec-trum(Fig.1)ledtotheidentificationofsubunitA(C-1→C-10)and
subunitB(C-2′→C-11′).Carefulinvestigationofdifferentspectral
datadeclaredthepresenceof2sp2carbonsincludinga trisubsti-tutedolefinicmoietyinthesubunitA.Thiswasprovedfromthe carbonsignalsresonatingatı136.8(qC)and124.8(CH).Thelatter carbonwasconnectedtotheprotonatıH5.14.Thesignal resonat-ingatı136.8wasattributedtoquaternarycarbonaspredictedfrom HSQCexperiment.HMBCexperiment(Fig.1)revealedacrosspeak fromH3-11(ı1.59)tothequaternarysp2carbon(ı136.8)beside theothersp2carbonandC-5(ı40.8).Thissupportedthelocationof theH3-11(ıH1.59/ıC16.1)asbeingdirectlylinkedtotheolefinic carbon(ı136.8).BothCOSYandHMBCdataconfirmedthelocation ofthedoublebondatC-6/C-7andalsoprovedthewholestructure ofsubunitA(C-1toC-10).InvestigationofthedifferentNMRdata (1HNMR,13CNMR,COSY,HSQCandHMBC)provedthe incorpo-rationof adoublebond andtwohydroxylated methineswithin subunitB. The COSYspectrumdeclared thespin–spin coupling betweentheprotonsresonatingatı3.98(H-3′)and4.12(H-2′)
whichare linkedtothecarbonsatı73.1(C-3′)and72.9(C-2′)
respectively.Thissupportedthelocationofthetwohydroxylsat C-2′andC-3′.TheHMBCdemonstratedacrosspeakfromtheproton
resonatingatı3.98(H-3′)totheamidiccarbonylatı177.2(C-1′)
andconfirmingtheassignmentofthesubstructureC-1′toC-3′.The
presenceofadoublebondatC-4′/C-5′wasconfirmedfromthe
pro-tonsignalsatı5.47(H-4′)and5.71(H-5′)whichcorrelatedtothe
signalsatı131.1and134.6respectivelyintheHSQCspectrum.The HMBCshowedcrosspeaksfromH-3′(ı3.98)toC-4′(ı131.1)and
hencesupportingthepresenceofolefinicmoietyatC-4′/C-5′.
ThelargecouplingconstantbetweenH-4′andH-5′(J=15.6Hz)
wasconsistentwithEconfiguration(Yoshikawaetal.,1996;Wang etal.,2009a,b).AsdeclaredfromCOSYandHMBCdata(Fig.1), thestructureofsubunitB(C-1′toC-11′)wasconfirmed.The
pres-enceofaglucosemoietywasconfirmedfromthesignalsresonating between62.6and104.7ppmin13CNMRspectrumalongwiththeir correspondingprotonsignalsdetectedbetween3.18–4.18ppmin 1HNMRspectrum.Thelinkbetweeneach protonand its corre-spondingcarbonwasproved fromHSQC.Thelargevalueofthe couplingconstant(J=7.8Hz)oftheanomericproton(H-1′′,ı4.27)
confirmeditsˇ-configuration(Pengetal.,2011;Renetal.,2009; Sheetal.,2009).Theattachmentoftheglucosemoietytothe sub-unit Awas confirmedfromtheHMBC cross-peaksof H-1/C-1′′.
Basedonthepreviousdiscussion,thestructureofcompound2was confirmed.Itisreortedhereforthefirsttimefromnaturalsource andwasgiventhegenericnamepenicillosideB.
Theantimicrobialactivityofcompounds1and2wasevaluated bydeterminingthegrowthinhibitionzone.Compound1revealed antifungalactivitytowardsC.albicansasitshowedinhibitionzone of23mm.Additionally,compound2wasactiveagainstS.aureus with19mminhibitionzoneandE.coli(20mm).Ontheotherhand, BothcompoundsshowedweakactivitywithIC50≥50g/mlwhen testedagainstHeLacells.
Conclusion
Investigationofthemarine-derivedfungusPenicilliumspecies yieldedtwonewcerebrosides,penicillosidesAand B(1and2). Compound1revealedantifungalactivitytowardsC.albicans. Addi-tionally,compound2showedantibacterialactivityagainstS.aureus andE.coli.Ontheotherhand,Bothcompoundsshowedweak activ-itywithIC50≥50g/mlwhentestedagainstHeLacells.
Authors’contribution
manuscriptandSSAMperformedtheexperiments,analyzedthe dataandwrotethemanuscript.
Conflictsofinterest
Theauthorsdeclarenoconflictsofinterest.
Acknowledgement
ThisprojectwassupportedbytheNSTIPstrategictechnologies programintheKingdomofSaudiArabia—ProjectNo. (11-BIO1556-03). The authors also, acknowledge with thanks Science and TechnologyUnit,KingAbdulazizUniversityfortechnicalsupport.
References
Abo-Kadoum,M.A.,Abo-Dahab,N.F.,Awad,M.F.,Abdel-Hadi,A.M.,2013. Marine-derivedfungus,PenicilliumaurantiogriseumAUMC9757:aproducerofbioactive secondarymetabolites.J.BasicAppl.Mycol.4,77–83.
Bugni,T.S.,Ireland,C.M.,2004.Marine-derivedfungi:achemicallyandbiologically diversegroupofmicroorganisms.Nat.Prod.Rep.21,143–163.
Blunt,J.W.,Copp,B.R.,Keyzers,R.A.,Munro,M.H.G.,Prinsep,M.R.,2013.Marine naturalproducts.Nat.Prod.Rep.30,237–323.
Boyd,M.R.,Paull,K.D.,Boyd,M.R.,Paull,K.D.,1995.Somepracticalconsiderations andapplicationsoftheNationalCancerInstituteinvitroanticancerdrug dis-coveryscreen.DrugDev.Res.34,91–109.
Devi,P.,Rodrigues,C.,Naik,C.G.,D’Souza,L.,2012.Isolationandcharacterization ofantibacterialcompoundfromamangrove-endophyticfungus,Penicillium
chrysogenumMTCC5108.IndianJ.Microbiol.52,617–623.
Elkhayat,E.S.,Mohamed,G.A.,Ibrahim,S.R.M.,2012.Activityandstructure elucida-tionofceramides.Curr.Bioact.Compd.8,370–409.
Fenical,W.,1993.Chemicalstudiesofmarinebacteria:developinganewresource. Chem.Rev.93,1673–1683.
Gao, H., Zhou, L., Li, D., Gu, Q., Zhu, T.J., 2013. New cytotoxic metabolites fromthemarine-derivedfungusPenicilliumsp.ZLN29.Helv.Chim.Acta96, 514–519.
Hall,T.A., 1999. BioEdit:a user-friendlybiological sequencealignment editor andanalysisprogramforwindows95/98/NT.NucleicAcidsSymp.Ser.41, 95–98.
Li,Q.,Wang,G.,2009.DiversityoffungalisolatesfromthreeHawaiianmarine sponges.Microbiol.Res.164,233–241.
Peng,X.P.,Wang,Y.,Sun,K.,Liu,P.P.,Yin,X.,Zhu,W.M.,2011.Cerebrosidesand 2-pyridonealkaloidsfromthehalotolerantfungusPenicilliumchrysogenumgrown inahypersalinemedium.J.Nat.Prod.74,1298–1302.
Qi,S.H.,Xu,Y.,Xiong,H.R.,Qian,P.Y.,Zhang,S.,2009.Antifoulingand antibacte-rialcompoundsfromamarinefungusCladosporiumsp.F14.WorldJ.Microbiol. Biotechnol.25,399–406.
Ren,S.C.,Liu,Z.L.,Ding,X.L.,2009.Isolationandidentificationoftwonovelflavone glycosidesfromcornsilk(Stigmamaydis).J.Med.PlantsRes.3,1009–1015. She,G.,Guo,Z.,Lv,H.,She,D.,2009.NewflavonoidglycosidesfromElsholtziarugulosa
Hemsl.Molecules14,4190–4196.
Singh,R.P.,Jain,D.A.,2011.AnticandidalpotentialofCrinumasiaticumleavesextract againstselectedoralandvaginalCandidapathogens.AsianJ.Biochem.Pharm. Res.1,283–291.
Skehan,P.,Storeng,R.,Scudiero,D.,Monks,A.,McMahon,J.,Vistica,D.,Warren,J.T., Bokesch,H.,Kenney,S.,Boyd,M.R.,1990.Newcolorimetriccytotoxicityassay foranticancer-drugscreening.J.Natl.CancerInst.82,1107–1112.
Subramani,R.,Kumar,R.,Prasad,P.,Aalbersberg,W.,2013.Cytotoxicand antibac-terialsubstancesagainstmulti-drugresistantpathogensfrommarinesponge symbiont:citrinin,asecondarymetaboliteofPenicilliumsp.AsianPac.J.Trop. Biomed.3,291–296.
Sun,Y.,Takada,K.,Takemoto,Y.,Yoshida,M.,Nogi,Y.,Okada,S.,Matsunaga,S., 2012.Gliotoxinanaloguesfromamarine-derivedfungus,Penicilliumsp.and theircytotoxicandhistonemethyltransferaseinhibitoryactivities.J.Nat.Prod. 75,111–114.
Tamura,K.,Peterson,D.,Peterson,N.,Stecher,G.,Nei,M.,Kumar,S.,2011.MEGA5: molecularevolutionarygeneticsanalysisusingmaximumlikelihood, evolution-arydistance,andmaximumparsimonymethods.Mol.Biol.Evol.28,2731–2739. Thompson,J.D.,Gibson,T.J.,Plewniak,F.,Jeanmougin,F.,Higgins,D.G.,1997.The CLUSTALXwindowsinterface:flexiblestrategiesformultiplesequence align-mentaidedbyqualityanalysistools.NucleicAcidsRes.25,4876–4882. Valgas,C.,deSouza,S.M.,Smània,E.F.A.,Smânia,J.,2007.Screeningmethods
todetermineantibacterialactivityofnaturalproducts.Braz.J.Microbiol.38, 369–380.
Wang,W.L.,Wang,Y.,Tao,H.W.,Peng,X.P.,Liu,P.P.,Zhu,W.M.,2009a.Cerebrosides ofthehalotolerantfungusAlternariaraphaniisolatedfromaseasaltfield.J.Nat. Prod.72,1695–1698.
Wang,Y.N.,Tian,L.,Hua,H.M.,Lu,X.,Sun,S.,Wu,H.H.,Pei,Y.H.,2009b.Twonew compoundsfromthebrothofthemarinefungusPenicilliumgriseofulvumY19-07. J.AsianNat.Prod.Res.11,912–917.
White,T.J.,Bruns,T.,Lee,S.,Taylor,J.,1990.Amplificationanddirectsequencing offungalribosomalRNAgenesforphylogenetics.In:Innis,M.A.,Gelfand,D.H., Sninsky,J.J.,White,T.J.(Eds.),PCRprotocols:Aguidetomethodsandapplication. AcademicPress,SanDiego,CA,USA,pp.315–322.