molecules
ISSN 1420-3049
© 2008 by MDPI www.mdpi.org/molecules
Full Paper
Synthesis and Total
1H- and
13C-NMR Assignment of Cephem
Derivatives for Use in ADEPT Approaches
Lorena Blau 1, Renato Farina Menegon 1, Elizabeth Igne Ferreira 2, Antonio Gilberto Ferreira 3, Elisangela Fabiana Boffo 3, Leila Aley Tavares 3, Vladimir Constantino Gomes Heleno 3,4,* and Man-Chin Chung 1,*
1
Departamento de Fármacos e Medicamentos, Faculdade de Ciências Farmacêuticas, Universidade Estadual Paulista, CP 502 – 14801-902, Araraquara, SP, Brazil; E-mails:
[email protected]; [email protected]
2
Departamento de Farmácia, Faculdade de Ciências Farmacêuticas, Universidade de São Paulo, CP 66083 – 05389-970, São Paulo, SP, Brazil; E-mail: [email protected]
3
Departamento de Química, Centro de Ciências Exatas e Tecnológicas, Universidade Federal de São Carlos, CP 676 – 13565-905, São Carlos, SP, Brazil; E-mails: [email protected];
[email protected]; [email protected]
4
Núcleo de Pesquisas em Ciências Exatas e Tecnológicas – Universidade de Franca, CP 82 – 14404-600, Franca, SP, Brazil
* Authors to whom correspondence should be addressed; E-mail for V.C.G.H.:
[email protected]; Phone: +55 16 37118871; Fax: +55 16 37118878; E-mail for M-C.C.: [email protected]; Phone: +55 16 33016962; Fax: +55 16 33016960.
Received: 12 March 2008; in revised form: 6 April 2008 / Accepted: 7 April 2008 / Published: 10 April 2008
5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid-3-[methyl 4-(6-methoxyquinolin-8-ylamino)pentylcarbamate]-8-oxo-7-[(2-thienyloxoacetyl)amino]- 5-dioxide (7) are also described, together with their total 1H- and 13C-NMR assignments.
Keywords: ADEPT, prodrug, cancer, cephalosporin, 1H, 13C and 2D NMR.
Introduction
About one third of the world’s population is currently at serious risk of contracting malaria. It is estimated that approximately 2-3 million people die from malaria every year, and in the sub-Saharan part of Africa alone about half million children below the age of five die of malaria in this same time period [1]. Treating malaria is becoming more difficult because of the spreading resistance of the parasite to standard antimalarial drugs, especially to chloroquine [2]. Most of the drugs currently employed in antimalarial chemotherapy are particularly active against the asexual blood forms of the parasite, responsible for the clinical symptoms of the disease [3]. With the rapid spread of drug-resistant Plasmodium falciparum strains, the development of safe and effective antimalarial drugs that prevent transmission and cure patients has become an important strategy toward an effective control of the disease [3].
In contrast with the asexual blood forms of Plasmodium, the sexual form of the parasite is a much less explored life-cycle target. Nowadays, primaquine is the only available transmission-blocking antimalarial drug that displays marked activity against gametocytes of all species of the parasite responsible for human malaria, including chloroquine-resistant P. falciparum. However, the use of primaquine is limited by its extensive metabolic conversion into carboxyprimaquine and its toxic effects, which include hemolytic anemia, particularly in the case of patients deficient in glucose-6-phosphate dehydrogenase [4,5].
As a way to solve problems such as low drug concentration in target cells, systemic toxicity [6], lack of selectivity for target cells over normal cells, and the appearance of drug-resistant cells, new drug delivery approaches have been the focus of much research in recent years [7], particularly for the cancer chemotherapy. One such approach is known as ADEPT (antibody-directed enzyme-prodrug therapy), and it consists of a promising two-step strategy, which should increase drug selectivity toward the target cells.
Cephalosporins are highly versatile carriers in the construction of enzyme-activated prodrugs, and they find potential application in ADEPT approaches. The prodrugs, which consist of cephalosporins substituted at the C-3’ position, are activated after antibody-beta-lactamase conjugate hydrolysis, which in turn releases the active drug and drives it toward the target [8]. This approach has been tested for anticancer therapy and is being currently applied to African trypanosomiasis [9].
Figure 1. Cephalosporin carrier for the ADEPT approach.
The second step involves administration of a prodrug with a specific substrate of the conjugated enzyme (carrier) covalently attached to a cytotoxic drug, so that the prodrug will be selectively activated and released on tumor cells. Cephalosporins were chosen as candidate prodrug carriers because of the readily controlled release of cytotoxic agents covalently attached to these compounds (Figure 1).
In this work, we present the synthesis of a cephalosporin-derivative carrier for use in ADEPT, to which any drug with a terminal amino group could be added. The synthesis of a prodrug from the cephalosporin derivative containing the atimalarial drug primaquine attached to it is also described. This prodrug could be used in malaria therapy, thus reducing the toxic effect associated with primaquine.
Detailed NMR data for the carriers and prodrugs used in the ADEPT approaches are considerably scarce in the literature, but particularly useful for this kind of research. Understanding what occur in each step of the ADEPT process is only possible if elucidation of the substances involved in these steps, namely the carrier, the drugs, the prodrugs and even the generated metabolites, is carried out. Therefore, a detailed analysis of the carrier 5, the intermediate 6 and the prodrug 7 was performed as part of our interest in structural elucidation by NMR techniques [10,11], and the total 1H- and 13C- NMR assignments for 5, 6 and 7, including 1H-NMR, 13C {1H}-NMR and 2D NMR (gCOSY, gHSQC and gHMBC) experiments, are presented.
Results and Discussion
Synthesis
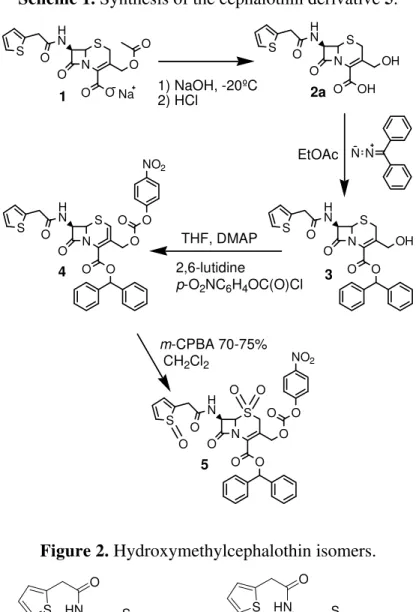
Scheme 1. Synthesis of the cephalothin derivative 5. N S O O O O H N O S Na N S O OH OH O H N O S
1 1) NaOH, -20ºC2) HCl 2a
N N EtOAc N S O O OH O H N O S 3 THF, DMAP 2,6-lutidine
p-O2NC6H4OC(O)Cl
N S O O O O H N O S O O O NO2 4
m-CPBA 70-75%
CH2Cl2
N S O O O O H N O
S O O
NO2
O O
O 5
Figure 2. Hydroxymethylcephalothin isomers.
N O HN O S S O OH OH N O HN O S S O OH OH 2a 2b
A strategic yet difficult step in the synthesis of cephalosporin-based prodrugs is a deacetylation process for the further drug attachment. Cephalosporin deacetylation by acid hydrolysis gives a γ -lactone compound, whereas alkaline hydrolysis results in opening of the lactam ring (Scheme 2). Both methods are unable to yield the prodrug that is employed in ADEPT [13].
This deacetylation can be achieved by enzymatic cleavage of the immobilized esterase [14], but this method is very difficult. Therefore, we carried out deacetylation by a convenient method developed by Mobashery for sodium cephalothin (1) [15], in which the special conditions of temperature (-20 ºC) and reaction time (135 seconds) are very important for the success of this reaction. We observed that when the reaction product is frozen and lyophilized, the degradation of 2a
does not occur.
reagents or methods which lead to lactonization should be avoided, so the preferred esterification reagents are aryldiazoalkanes, particularly aryldiazomethanes. Alternatively, the ester may be prepared by reacting an aralkyl chloride or bromide with an alkali metal salt; e.g., the sodium salt, of the 3-hydroxymethylcephemoic acid analogue. Examples of esters that may be prepared by this latter method are the 4-triphenylmethyl esters and 4-benzyl esters [16].
Scheme 2. Cephalothin degradation.
HCl, 0 ºC NaOH, 0 ºC
(1)
γ-lactone β-lactam ring opened
N HN
O
S
O S
O O
-O
O Na+
N HN
O
S
O S
O O
N HN
O
S S
O O-Na+
-O
O Na+
The protection of the carboxylic group of cephalosporins is necessary for subsequent drug attachment to these compounds. Jungheim performed isomerization of hydroxymethylcephalothin (2a) to isomer 2b (Figure 2), a more soluble product, before attaching the protection [12]. We carried out the protection of hydroxymethylcephalothin (2a) in good yield without this isomerization, so precautions against isomerization are not necessary and one step can be avoided.
Compound 3 was then reacted with p-nitrophenylchloroformate, leading to the attachment of a good leaving group under basic conditions, which should facilitate drug attachment [17].Moreover, the resulting carbamate moiety, to which the drug can be attached, should provide an appropriate and efficient environment for drug release from the cephem nucleus during the ADEPT process [18].
The antimalarial drug primaquine was then successfully attached to carrier 5, leading to the cephalothin-primaquine prodrug 7, as shown in Scheme 3.
Scheme 3. Preparation of the cephalothin-primaquine prodrug (7).
N S
O O
O O
O H N
O O S
O O
NO2 O
N S
O O
O HN
O H N
O O S
O O
N H N
O O
primaquine, THF
room temperature, 24h
5
6
N S
OH O O HN
O H N
O O S
O O
N H N
O O
7
TFA, CH2Cl2, anisole 0 ºC
The condensation of 5 with primaquine in THF provided the desired carbamate 6 in good yield, and the final compound 7 was easily obtained through deprotection with trifluoroacetic acid (TFA) and anisole in dichloromethane.
NMR Assignments
Literature NMR data on substances like 5 and 7 are rare, incomplete and little detailed. However, total characterization cannot be completely achieved without the complete assignment of NMR signals. Therefore, we decided to carry out the total NMR characterization of these compounds so as to provide enough data for their prompt structural determination in further works.
Compound 5 was analyzed by the 1H-NMR, 13C-NMR{1H}, and gHMBC techniques. The 1H- and
13
C-NMR data assignments were carefully undertaken by detailed analysis of the 1H-NMR spectrum. Expansions were made for all signals, which allowed the measurement of several J values. Analysis of the multiplicities and their coupling constants was useful to verify and confirm some 1H/1H correlations.
Table 1. 1H- and 13C-NMR chemical shifts for compound 5.
N S
O O
O N
O
O NO2
O
O S
O H
1 2
3
4 5 6 7
8 9 10 11 12 13 14
15 16
1'
2' 3' '4 5'
6' 1''
2'' 3'' 4''a
5''
6'' 4''b
Position δ13C (ppm) δ1H (ppm); (int., mult.); J (Hz) gHMBC
2 45.4 a – 4.08; (1H, d); J=18.6 C3; C4; C6 b – 3.71; (1H, d); J=18.6
3 120.1 --- ---
4 125.3 --- ---
6 66.6 4.99; (1H, d); J=4.5 C8
7 58.2 5.98; (1H, dd); J=8.3; J=4.5 C6; C8; C10
8 164.5 --- ---
9 --- 8.47; (1H, d); J=8.3 C7; C8; C10
10 170.2 --- ---
11 35.7 a – 3.92; (1H, d); J=15.5 C10; C12; C13 b – 3.83; (1H, d); J=15.5
12 136.7 --- ---
13 126.9 7.29; (1H, d); J=5.5 ---
14 126.7 7.28; (1H, dd); J=6.7; J=5.5 ---
15 125.1 7.54; (1H, d); J=6.7 ---
1’ 67.4 a – 5.30 ; (1H, d); J=13,0 C2; C3; C4; C2’ b – 4.91; (1H, d); J=13,0
2’ 151.5 --- ---
3’ 155.1 --- ---
4’ 122.4 7.52; (2H, d); J=9.0 C3’; C5’; C6’ 5’ 125.4 8.31; (2H, d); J=9.0 C3’; C5’; C6’
6’ 145.2 --- ---
1” 159.7 --- ---
2” 79.3 6.96; (1H, s) C1’’; C3’’; C4’’
3” 139.6 --- ---
4” 126.6 a – 7.44; (2H, d); J=7.4 C2’’ b – 6.98; (2H, d); J=3.5
5” 128.5 7.34; (4H, dd); J=7.4; J=6.7 C3’’; C6’’
6” 128.4 7.38; (2H, dd); J=6.7; J=3.5 ---
Table 2. 1H- and 13C-NMR chemical shifts for compound 6. N S O N O N O O O S O H 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 1' 2' 1'' 2'' 3'' 4''a 5'' 6'' O O O N N H H O 3' 4' 5' 6' 7' 8' 9' 10' 11' 12' 13' 14' 15' 16' 17' 18' 19' 20' 4''b
Position δ
1H (ppm), (int., mult.), J
(Hz) δ
13C (ppm) gHSQC gHMBC
2 a – 3.62; (1H, d); J=18.8 45.6 C2 C3; C4; C6; C1’ b – 2.94; (1H, d); J=18.8
3 --- 123.4 --- ---
4 --- 124.6 --- ---
6 4.17 – 4.26; (1H, m) 66.7 C6 C8
7 5.81 – 5.96; (1H, m) 59.0 C7 C6; C8; C10
8 --- 164.5 --- ---
9 6.99; (1H, d); J=9.8 --- --- C7; C10
10 --- 170.5 --- ---
11 3.72; (2H, s) 37.2 C11 C10; C11; C12
12 --- 135.0 --- ---
13
14 6.83 – 6.92; (2H, m)
127.2
127.4 C13; C14 C12; C15 15 7.08 – 7.40; (1H, m) 125.9 C15 ---
1’ a – 5.05; (1H, d); J=14.3 63.4 C1’ C2; C3; C4; C2’ b – 4.52; (1H, d); J=14.3
2’ --- 156.0 --- ---
4’ 2.99 – 3.13; (2H, m) 41.2 C4’ ---
5’, 6’ 1.38 – 1.66; (4H, m) 26.6; 33.9 --- C4’; C5’; C6’; C7’; C8’ 7’ 3.43 – 3.55; (1H, m) 47.9 --- C5’; C6’
8’ 1.18; (3H, d); J=6.0 20.6 C8’ C6’; C7’
10’ --- 144.9 --- ---
11’ 6.25; (1H, brs) 92.0 C11’ C12’; C13’; C19’
12’ --- 159.6 --- ---
13’ 6.19; (1H, brs) 97.1 C13’ C11’; C12’; C19’
14’ --- 130.1 --- ---
15’ 7.83; (1H, d); J=8.1 135.1 --- ---
16’ 7.07 – 7.41; (1H, m) 122.1 --- ---
17’ 8.43; (1H, dd); J=5.8; J=1.6 144.4 C17’ C16’; C19’
19’ --- 135.3 --- ---
20’ 3.79; (3H, s) 55.5 C20’ C12’; C20’
1’’ --- 160.1 --- ---
2’’ 6.81; (1H, s) 80.4 C2’’ C1’’; C3’’; C4’’a; C4’’b
3’’ --- 139.2 --- ---
4’’a, 4’’b 5’’, 6’’
7.08 – 7.40; (10H, m)
127.7; 127.8 128.6; 128.8; 128.3; 128.4
--- ---
Table 3. 1H- and 13C-NMR chemical shifts for compound 7. N S O N OH N O O O S O H 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 1' 2' 1'' O O O N N H H O 3' 4' 5' 6' 7' 8' 9' 10' 11' 12' 13' 14' 15' 16' 17' 18' 19' 20'
Position δ1H (ppm); (int., mult.); J (Hz) δ13C (ppm) gCOSY gHSQC gHMBC
2 3.75 – 3.94; (2H, m) 45.2 --- C2 C3; C4; C6
3 --- 119.7 --- --- ---
4 --- 125.3 --- --- ---
6 4.88; (1H, d); J=4.8 66.2 H7 C6 C8
7 5.80; (1H, dd); J=8.4; J=4.8 58.0 H6; H9 C7 C6; C8; C10
8 --- 164.1 --- --- ---
9 8.42; (1H, d); J=8.4 --- H7 --- C7; C8; C10
10 --- 170.0 --- --- ---
11 3.75 – 3.94; (2H, m) 35.6 --- C11 C10; C12; C13
12 --- 136.8 --- --- ---
13; 14 6.91 – 6.97; (2H, m) 126.4; 126.7 H15 C13; C14 C12; C15 15 7.36; (1H, dd); J=4.8; J=1.8 125.1 H13; H14 C15 C12; C13
1’ a – 5.11; (1H, d); J=13.1
b – 4.52; (1H, d); J=13.1 62.7 H1’a; H1’b C1’ C2; C3; C4; C2’
2’ --- 155.8 --- --- ---
3’ 7.29; (1H, t); J=5.9 --- H4’ --- ---
4’ 2.98; (2H, d); J=5.9 40.3 H3’; H5’; H6’ C4’ --- 5’; 6’ 1.41 – 1.70; (4H, m) 26.1; 33.2 H4’; H7’ C5’; C6’ C8’
7’ 3.55 – 3.66; (1H, m) 47.0 H8’ C7’ ---
8’ 1.19; (3H, d); J=6.3 20.1 H7’ C8’ C6’; C7’
10’ --- 144.5 --- --- ---
11’ 6.48; (1H, d); J=2.6 91.7 H13’ C11’ C12’; C13’; C15’
12’ --- 159.0 --- --- ---
13’ 6.25; (1H, d); J=2.6 96.3 H11’ C13’ C12’; C15’
14’ --- 129.6 --- --- ---
15’ 8.08; (1H, dd); J=8.4; J=1.5 135.0 H16’ C15’ C11’; C15’; C17’ 16’ 7.43; (1H, dd); J=8.4; J=4.2 122.1 H15’; H17’ C16’ C14’; C17’
17’ 8.53; (1H, dd); J=4.2; J=1.5 144.1 H16’ C17’ C15’; C16’
19’ --- 134.3 --- --- ---
20’ 3.75 – 3.94; (3H, m) 55.0 --- C20’ C12’; C20’
1’’ --- 162.0 --- --- ---
Conclusions
A new cephalosporin carrier and a new correlated prodrug were successfully synthesized. The carrier itself is now available for use with a wide range of drugs. The preparation of the primaquine prodrug shows that several prodrugs can be developed. The NMR study undertaken for the carrier, the prodrug and the intermediate product furnished a considerable amount of new NMR data. These data can be used as a tool to verify and prove structural changes in future works.
Experimental
General
Petroleum ether, ethyl acetate, dichloromethane and THF were dried with anhydrous sodium sulfate and distilled at atmospheric pressure. Flash chromatography was carried out on Merck Kieselgel 60 silica gel (230-400 mesh). Melting points were measured in an Eletrothermal 9200 apparatus and are uncorrected. All glassware was oven-dried, assembled hot, and cooled under a stream of dry nitrogen before use. Infrared spectra were recorded with a Shimadzu FTIR-8300 spectrophotometer. Elemental analysis was performed in a Perkin-Elmer 240 A analyzer.
NMR Experiments
All NMR experiments were performed on a Bruker DRX400 instrument (9.4 T) operating at 400.13 MHz for 1H and at 100.58 MHz for 13C. The 1H-NMR and 2D-NMR (gCOSY, gHSQC and gHMBC) experiments were undertaken with an inverse 5 mm probehead (BBI), and the 13C {1 H}-NMR experiments were carried out with a direct 5 mm probehead (BBO). The temperature of the sample was always 298 K and the concentrations ranged from 15 to 35 mg·mL-1 in DMSO-d6. TMS
was used as internal reference.
Synthesis
Cephalothin derivative 5
Cephalothin sodium salt (1) was provided by EMS/Sigma Pharma Group and was readily deacylated by basic hydrolysis at – 20 ºC [6]. Hydroxymethylcephalothin (2a) was reacted with diphenyldiazomethane, a carboxylic blocking agent that was prepared by oxidation of benzophenone hydrazone (see below), to give compound 3. The latter was substituted at the C-3 hydroxyl group with p-nitrophenylchloroformate, using 4-DMAP and 2,6-lutidine as catalysts, to give a mixture of isomers
4. Oxidation of 4 with m-chloroperbenzoic acid (m-CPBA) provided the derivative cephalothin 5
Diphenyldiazomethane
A mixture of benzophenone hydrazone (9.8 g; 0.05 mol) and yellow mercuric oxide (11 g; 0.05 mol) in dry petroleum ether (50 mL) was placed in a pressure bottle at room temperature for six hours. This mixture was filtered and some insoluble material was removed. The filtrate was evaporated, giving a dark red oil at room temperature. Freezing this oil with dry ice and then allowing it to warm to room temperature gave dark red crystals. M.p. 29-30 ºC; IR (KBr, cm-1): 2037 (diazo) and 1593, 1495 (aromatic ring).
3-Hydroxymethyl-8-oxo-7-[(2-thienylacetyl)-amino]-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid (2a)
An aqueous solution (15 mL) of sodium cephalothin (1, 3.0 g, 7.48 mmol) was added at 0 ºC to NaOH 19% (30 mL) that had been cooled to –20 ºC. The reaction mixture was stirred at –10 ºC for 135 seconds and then quenched by the rapid addition of glacial acetic acid (10 mL) at room temperature. The reaction vessel was then transferred to an ice-water bath and the pH of the mixture was immediately lowered to 1.5 by adding concentrated HCl, which had previously been cooled to –20 ºC. The white solid was filtered, washed twice with cold water, suspended in water, frozen and lyophilized to give 2a as a white powder (2.07 g, 80%); m.p. 210 ºC (dec.); IR (KBr, cm-1): 3413 (OH), 1760 (β-lactam ring), 1719 (COOH) and 1655 (amide).
3-Hydroxymethyl-8-oxo-7-[(2-thienylacetyl)amino]-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic aciddiphenylmethyl ester (3)
A solution of diphenyldiazomethane (3.24g, 16.68 mmol) in dry ethyl acetate (50 mL) was added dropwise to a mixture of hydroxymethylcephalothin (2a; 6.0g, 16.92 mmol) in dry ethyl acetate (250 mL). The reaction mixture was stirred at room temperature for four hours and then filtered. The organic layer was washed with cold aqueous saturated NaHCO3 (3 x 200 mL) and cold water (1 x 200
mL), and dried over anhydrous Na2SO4. The organic layer was concentrated in vacuum to give 3 as a
white powder (10.0 g, 52.44%); m.p. 165 – 168 ºC; IR (KBr, cm-1): 1755 (β-lactam ring), 1724 (ester), and 1666 (amide).
5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid-3-[[[(4’’-nitrophenoxy)carbonyl]oxy]methyl]-8-oxo-7-[(2-thienylacetyl)amino]-diphenylmethyl ester and 5-thia-1-azabicyclo[4.2.0]oct-3-ene-2-carboxylic acid-3-[[[(4’’-nitrophenoxy)carbonyl]oxy]methyl]-8-oxo-7-[(2-thienylacetyl)amino]- diphenylmethyl ester (4)
under vacuum, to give an oil. This oil was dissolved in dichloromethane (100 mL), washed with cold 0.2 N HCl (3 x 50 mL) and cold water (1 x 50 mL), dried over anhydrous Na2SO4, and purified by
flash column chromatography using 5% ethyl acetate in dichloromethane as eluent to give 9.0 g of 4
(69% yield). M.p. 69-71 ºC; IR (CHCl3, cm-1): 1780 (β-lactam ring), 1751 (carbonate), 1684 (amide)
and 1529 (NO2).
5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid-3-[[[(4’’-nitrophenoxy)carbonyl]oxy]methyl]-8-oxo-7-[(2-thienyloxoacetyl)amino]-diphenylmethyl ester-5-dioxide (5)
m-Chloroperbenzoic acid (70-75%; 0.65 g, 2.75 mmol) in dichloromethane (20 mL) was added dropwise to a solution of 4 (0.75 g, 1.1 mmol) in dichloromethane (20 mL) at 0 ºC, over a period of 15 min. The reaction mixture was stirred for 4 hours at 0 ºC and then washed with cold saturated aqueous NaHCO3 (4 x 50 mL) and cold saturated aqueous NaCl (1 x 50 mL). The organic layer was dried over
anhydrous Na2SO4 and concentrated under vacuum. The residue was triturated with methanol, to give
pure 5 (0.46g, 60%) as a white powder. M.p. 164-168 ºC; IR (KBr, cm-1): 1786 (β-lactam ring), 1767 (carbonate), 1655 (amide) and 1720 (ester); Anal. Calcd. for C34H27N3O12S2: C, 55.66; H, 3.71; N,
5.73. Found: C, 55.70; H, 3.72; N, 5.71. The 1H- and 13C-NMR chemical shifts were assigned and all the values are listed in Table 1.
5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid-3-[methyl 4-(6-methoxyquinolin-8-ylamino)-pentylcarbamate]-8-oxo-7-[(2-thienyloxoacetyl)amino]-diphenylmethyl ester-5-dioxide (6)
Primaquine diphosphate (500 mg, 1.1 mmol) water solution (30 mL) was neutralized with 5% K2CO3 and extracted with ethyl ether (50 mL). The organic phase was evaporated and the residue was
dissolved in THF (10 mL). The obtained solution was added to compound 5 (220 mg, 0.3 mmol). After stirring for 24 h at room temperature, the target compound 6 was isolated by column chromatography on silica gel employing 30 to 50% ethyl acetate in dichloromethane as eluent. This procedure yielded 166 mg of 6 (195 mmol; 65%). M.p. 162 – 164 ºC; IR (KBr, cm-1): 1794 (β-lactam ring), 1718 (ester), 1695 (carbamate) and 1659 (amide). The 1H- and 13C-NMR chemical shifts were assigned and all the values are listed in Table 2.
5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid, 3-[methyl 4-(6-methoxyquinolin-8-ylamino)-pentylcarbamate]-8-oxo-7-[(2-thienyloxoacetyl)amino]- 5-dioxide (7)
Distilled anisole (1.0 mL) and freshly distilled CF3CO2H (TFA, 1.0 mL) were added to a solution
of 6 (200 mg, 0.23 mmol) in dry CH2Cl2 (10 mL) at -20 ºC. After 15 minutes, the reaction was warmed
Acknowledgements
The authors are grateful to FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo), CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) for financial support.
References
1. Vangapandu, S.; Sachdeva, S.; Jain, M.; Singh, S.; Singh, P. P.; Kaul, C. L.; Jain, R. 8-Quinolinamines conjugated with amino acids are exhibiting potent blood-schizontocidal antimalarial activities. Bioorg. Med. Chem. 2004, 12, 239-247.
2. Gomes, P.; Araújo, M. J.; Rodrigues, M.; Vale, N.; Azevedo, Z.; Iley, J.; Chambel, P.; Morais, J.; Moreira, R. Synthesis of imidazolidin-4-one and 1H-imidazo[2,1-a]isoindole-2,5(3H,9bH)-dione derivatives of primaquine: scope and limitations. Tetrahedron, 2004, 60, 5551-5562.
3. Araújo, M. J.; Bom, J.; Capela, R.; Casimiro, C.; Chambel, P.; Gomes, P.; Iley, J.; Lopes, F.; Morais, J.; Moreira, R.; De Oliveira, E.; Do Rosário, V.; Vale, N. Imidazolidin-4-one Derivatives of Primaquine as Novel Transmission-Blocking Antimalarials. J. Med. Chem, 2005, 48, 888-892. 4. Mihaly, G. W.; Ward, S. A.; Edwards, G.; Orme, M. L.; Breckenridge, A. M. Pharmacokinetics of
primaquine in man: identification of the carboxylic acid derivative as a major plasma metabolite. Br. J. Clin. Pharmacol. 1984, 17, 441-446.
5. Constantino, L.; Paixão, P.; Moreira, R.; Portela, M. J.; Rosário, V. E.; Iley, J. Metabolism of primaquine by liver homogenate fractions. Evidence for monoamine oxidase and cytochrome P450 involvement in the oxidative deamination of primaquine to carboxyprimaquine. Exp. Toxicol. Pathol. 1999, 51, 299-303.
6. (a) Tsalic, M.; Bar, S. G.; Beny, A.; Visel, B.; Haim, N. Severe Toxicity Related to the 5-Fluorouracil/Leucovorin Combination (The Mayo Clinic Regimen): A Prospective Study in Colorectal Cancer Patients. Am. J. Clin. Oncol. 2003, 26,103-106; (b) Extermann, M.; Chen, H.; Cantor, A. B.; Corcoran, M. B.; Meyer, J. Predictors of tolerance to chemotherapy in older cancer patients: a prospective pilot study. Eur. J. Cancer 2002, 38, 1466-1473.
7. (a) Blagosklonny, M. V. Drug-resistance enables selective killing of resistant leukemia cells: exploiting of drug resistance instead of reversal. Leukemia 1999, 13, 2031-2035; (b) Fracasso, P. M.; Westerveldt, P.; Fears, C. A.; Rosen, D. M.; Zuhowski, E. G.; Cazenave, L. A.; Litchman, M.; Egorin, M. J. Phase I Study of Paclitaxel in Combination With a Multidrug Resistance Modulator, PSC 833 (Valspodar), in Refractory Malignancies. J. Clin. Oncol. 2000, 18, 1124-1134.
8. Blau, L.; Menegon, R. F.; Chung, M. C. Pró-fármaco ativado por enzima, uma estratégia promissora na quimioterapia. Quim. Nova 2006, 29, 1307-1317.
10. Heleno, V. C. G.; Silva, R.; Pedersoli, S.; Albuquerque, S.; Bastos, J. K.; Silva, M. L. A.; Donate, P. M.; Silva, G. V. J.; Lopes, J. L. C. Detailed 1H and 13C NMR structural assignment of three biologically active lignan lactones. Spectrochim. Acta A 2006, 63, 234-239.
11. Heleno, V. C. G.; Crotti, A. E. M.; Constantino, M. G.; Lopes, N. P.; Lopes, J. L. C. Total assignment of 1H and 13C NMR data for the sesquiterpene lactone 15-deoxygoyazensolide. Magn. Reson. Chem. 2004, 42, 364-367.
12. Jungheim, L. N.; Shepherd, T. A.; Meyer, D. L. Synthesis of acylhydrazido-substituted cephems. Design of cephalosporin-vinca alkaloid prodrugs: substrates for an antibody targeted enzyme. J. Org. Chem. 1992, 57, 2334-2340.
13. Nomura, H.; Fugono, T.; Hitaka, T.; Minami, I.; Azuma, T.; Morimoto, S.; Masuda, T. Semisynthetic β-lactam antibiotics. 6. Sulfocephalosporins and their antipseudomonal activities. J. Med. Chem. 1974, 17, 1312-1315.
14. Suzuki, N.; Sowa, T.; Murakami, M. Process for preparing 7-aminocephalosporanic acid derivatives. US Pat. 4,079,180, 1978.
15. Mobashery, S.; Johnston, M. Reactions of Escherichia coli TEM β-lactamase with cephalothin and with C10-dipeptidyl cephalosporin esters. J. Biol. Chem. 1986, 261, 7879-7887.
16. Somerfield, G. A.; Wycombe, H.; Chagouri, H. Cephemoic acids and process for preparing same. US Pat. 3,532,694, 1970.
17. Letsinger, R. L.; Ogilvie, K. K. Use of p-nitrophenyl chloroformate in blocking hydroxyl groups in nucleosides. J. Org. Chem. 1967, 32, 296-300.
18. Alexander, R. P.; Bates, R. W.; Pratt, A. J.; Kraunsoe, J. A. E. AN-nitrosochloroethyl-cephalosporin carbamate prodrug for antibody-directed enzyme prodrug therapy (ADEPT). Tetrahedron 1996, 52, 5983-5988.
19. Kaiser, G. V.; Cooper, D. G.; Koehler, R. E.; Murphy, C. F.; Webber, J. A.; Wright, I. G.; Van Heyningen, E. M. Chemistry of cephalosporin antibiotics. XIX. Transformation of Δ2-cephem to
Δ3
-cephem by oxidation-reduction at sulfur. J. Org. Chem. 1970, 35, 2430-2433.
Sample Availability: No compound samples are available.