w w w. s b f g n o s i a . o r g . b r / r e v i s t a
Original
Article
Comparison
and
evaluation
of
two
methods
for
the
pesticide
residue
analysis
of
organophosphates
in
yerba
mate
Lucía
Pareja
a,
Silvina
Niell
a,
Zisis
Vryzas
b,1,
Joaquín
González
c,
María
Verónica
Cesio
a,c,
Euphemia
P.
Mourkidou
b,
Horacio
Heinzen
a,c,∗aPoloAgroalimentarioyAgroindustrial,DepartamentodeQuímicadelLitoral,CentroUniversitariodePaysandú,UniversidaddelaRepública,EstaciónExperimentalMarioCassinoni,
Ruta3,Km363,Paysandú,Uruguay
bAristotleUniversityofThessaloniki,PesticideScienceLaboratory,P.O.Box1678,54124Thessaloniki,Greece
cFarmacognosiayProductosNaturales,DepartamentodeQuímicaOrgánica,FacultaddeQuímica,UdelaR,GeneralFlores2124,11800Montevideo,Uruguay
a
r
t
i
c
l
e
i
n
f
o
Articlehistory:
Received16July2014 Accepted2February2015 Availableonline21March2015
Keywords:
Yerbamate Pesticideresidues QuEChERS MAE GC–FPD
a
b
s
t
r
a
c
t
MicrowaveAssistedExtractionandamodifiedCEN-QuEChERSmethodologywereevaluatedas extrac-tionandcleanupproceduresforthesimultaneousanalysisof42organophosphatepesticidesinyerba mate(Ilexparaguaiensis).Theobtainedextractswereanalyzedbygaschromatographyusingaflame photometricdetector.Linearity,recoverypercentages,relativestandarddeviations,detectionand quan-tificationlimitsandmatrixeffectsweredeterminedaccordingtoDG-SANCOguidelinesforbothmethods. At0.2and0.5mg/kgtheevaluatedmethodsshowedpercentagesrecoveriesbetween70and120%for mostoftheanalytes.UsingMicrowaveAssistedExtractionmethodology,33pesticideresiduescouldbe properlyanalyzedwhereasonly27couldbedeterminedwiththeproposedmodifiedQuEChERS.All rel-ativestandarddeviationwerebelow18%exceptforomethoateanddisulfotonsulfonewhenevaluated bythemodifiedQuEChERS.Thelimitsofdetectioninbothmethodologieswere0.2mg/kgformostof theanalyzedcompounds.TheaveragedetectionlimitforQuEChERSwas0.04mg/kg.For19ofthe ana-lytesdeterminedthroughMicrowaveAssistedExtractionthelowestvalidatedlevelwere0.004mg/kg. Signalsuppression/enhancementwasobservedformostofthepesticides,thusmatrix-matched calibra-tioncurveswereusedforquantification.TheMicrowaveAssistedExtractionandQuEChERSprocedures studiedcoulddetecttheorganophosphatepesticidesabovetheMRLfixedfor“mate”bytheEuropean Union.Theyhavebeensuccessfullyappliedforthedeterminationoforganophosphatepesticideresidues incommercialsamplesandthepositiveswereconfirmedthroughGC–(ITD)-MS.
©2015SociedadeBrasileiradeFarmacognosia.PublishedbyElsevierEditoraLtda.Allrightsreserved.
Introduction
IlexparaguariensisA.St.-Hil.,Aquifoliaceae,isanativetreefrom theRiodelaPlatabasininSouthAmerica.Ithasbeencultivated sincecolonialtimes.Nowadays,300,000tonsofprocessedleaves
areconsumedeach year,which areusedtoprepareaninfusion
calledMate,thenationalbeverageofUruguay,Argentina,southern Brazil,andParaguay.Theartofmatedrinkinghasbeendescribed
∗ Correspondingauthorat: PoloAgroalimentarioy Agroindustrial, Departa-mentodeQuímicadelLitoral,CentroUniversitariodePaysandú,Universidadde laRepública,EstaciónExperimentalMarioCassinoni,Ruta3,Km363,Paysandú, Uruguay.
E-mail:[email protected](H.Heinzen).
1Presentaddress:DemocritusUniversityofThrace,FacultyofAgricultural
Devel-opment,LaboratoryofAgriculturalPharmacology&Ecotoxicology,193,Pantazidou str.68200,N.Orestiada,Greece.
byPérezParadaetal.(2010),Jacquesetal.(2007)andVázquez
andMoyna(1986).Thistraditionalbeverageisreputedtohavea
characteristicbittertasteandhepatoprotective,choleretic, hypoc-holesteremic, antioxidant, antirheumatic, diuretic and lipolitic properties(Filipetal.,2001).
Asanyothercrop,yerbamateisattackedduringfarmingby pests,especially mites,leaf-eatingbeetlesand caterpillars forc-ing theuseof organophosphate insecticides, that left pesticide residues.AsyerbamatehasbeenbeingsoldsteadilyinEuropealone orincombinationwithotherherbsasenergyteaorasaweight reductionaid(Andradeetal.,2012;Hecketal.,2007)the Euro-peanUnionhasestablishedMRLofpesticideresiduesontheleaves
(EuropeanCommission,2005).
Yerbamateisacomplexmatrixforpesticideresiduesanalysis dueitschemicalcomposition(naturalpigments,lipids,vitamins andsecondarymetabolites:polyphenols,saponins,andxanthines likecaffeineand theobromine)(Hecketal., 2007;Vázquez and
http://dx.doi.org/10.1016/j.bjp.2015.02.001
Moyna, 1986), and only few studieshave beenreported(Pérez
Parada et al., 2010; Jacques et al., 2006). Particularly, caffeine
andsaponinsareco-extractedwithpesticidesastheyhave sim-ilar physicochemical properties. Large amounts of caffeine and saponinscontaminatetheinjectorandthedetectoroftheGC sys-tem,interferingwiththedeterminationofpesticideresidues(Xu
etal.,2011;PérezParadaetal.,2010).Thegaschromatographic
separation of pesticides has been reviewed. Several analytical
strategies and column types have been proposed for pesticide
residueanalysisinmatricessuchastea,tobaccoandherbs(Liuand
Min,2012;Khanetal.,2014).
Theactualtrendforpesticideresiduesdeterminationattrace levelsseeksforvalidatedanalyticalmethodswithshorteranalysis timeandhighersamplethroughput(Chenetal.,2011).Considering matea“tea-like”matrix,thereareseveralmethodologiesreported fortheanalysisofpesticideresiduesinmadetea,teainfusionand spentleaves.Thesemethodsinclude,forexample,extractionwith differentsolventslikeethylacetate(EtOAc),cyclohexaneor ace-tonitrile,combinedwithdifferentcleanupprocedures;suchasgel permeation,andsolidphasecleanup,eitherdispersiveorusing car-tridges,followedbyliquidorgaschromatographyanalysis,coupled tomassdetectors(Huangetal.,2007,2009;Kanraretal.,2010).
Lozanoetal.(2012)andCajkaetal.(2012),describedthe
applica-tionofamodifiedQuEChERSforthedeterminationofpesticides indifferenttypesofteas.TheQuEChERSapproachisavery flex-ibleoneasitisatemplate toadapt theprocedureaccordingto analyteproperties,matrixcomposition,equipmentandanalytical techniquesavailableinthelaboratory(Anastassiadesetal.,2003). QuEChERSbasedmethodshavebeenusedtoassesfoodsafetyand environmentalsustainability.SeveralreportsonQuEChERS appli-cationsinherbshavebeendevelopedbuttherearenoreportson QuEChERSfortheanalysisofpesticideresiduesinyerbamateleaves
(Sadowska-Rocieketal.,2013;Attallahetal.,2012;Lozanoetal.,
2012;Chenetal.,2011,2012a,b;Nguyenetal.,2010;Haywardetal.,
2013).
Some other methodologies employing pressurized liquid
extraction,dispersiveliquid–liquidmicroextractionanddispersive solidphaseextractionhavebeendescribedintheliteratureforthe analysisofpesticideresiduesintea(Nguyenetal.,2010;Moinfar
etal.,2009;Choetal.,2008).Microwaveassistedextraction(MAE)
hasbeenassayedasextractionandclean upprocedurein food
matrices(Vryzasetal.,2007;Papadakisetal.,2006;Vryzasetal., 2002),butthereisnoreportforMAEinherbalteas.Itsmain advan-tagesarelowsolventconsumption,shortextractiontime,andhigh levelof automationwithhighextractionefficiency(Niell etal.,
2011;Papadakisetal.,2006).
ThepresentworkcomparesMAEandQuEChERSperformance
forpesticideresiduesanalysisofyerbamateleaves.
Materialsandmethods
Analyticalstandards andpesticidegrade solventswerefrom
Promochem(Wesel,Germany),Riedel-deHáën(Seelze,Germany)
and Merck (Darmstadt, Germany). Anhydrous magnesium
sul-phate(MgSO4), Graphitized CarbonBlack (GCB) and ENVI-carb
SPE, cartridge and PSA (primary–secondary amine) were from
Sigma–Aldrich(Madrid,Spain).Sep-Paksilicacartridgeswerefrom WatersCorporation(Milford,MA,USA),PSAsodiumcitratedibasic sesquihydrateandsodiumcitratetribasicdihydrateweresupplied fromSupelco(Bellefonte,PA,USA).
Stocksolutionsofindividualanalytesat1mg/mlwereprepared
in EtOAc; three mixed standard stock solutions were prepared
and seriallydilutedwithEtOAc toproducea series ofworking standard solutions of 0.001–20mg/l. The latter solutions were usedfortheconstructionofcalibrationcurvesandthepreparation
of the fortified samples. Stock solutions were stored in deep
freeze(−23◦C),whiletheworkingstandardsolutionswerestored
refrigerated and renewed at weekly intervals. Matrix-matched
calibrationsolutions(0.05–4g/ml)wereprepareddrying0.2ml
yerbamate extractundera N2 streamand fortifiedwith0.2ml workingstandardsolutionsofpesticidesatvariousconcentrations. Thesematrix-matchedsolutionswereusedtopreparecalibration curves,toevaluatethelinearrange,andtocalculaterecoveries.
Apparatus
TheMSP1000laboratorymicrowavesystem(CEM,Matthews,
NC)equippedwith12vesselcarouselwithtemperatureand
pres-sure sensors, operated in the closed mode was used for the
microwaveassistedextraction(MAE)ofyerbamateleaves. PTFE-linedextractionvesselswereused.
PesticideresiduesanalysiswasperformedinaThermoFisher Scientific,modelFinniganTraceGC(Rodano,Milan,Italy),gas chro-matographequippedwithaflamephotometricdetector(FPD),an
autosampler(modelAS3000), and aProgrammed Temperature
Vaporizer(PTV)(initialtemperaturewas60◦C(holdfor1.5min)
thenincreasedto220◦Cattherateof5◦C/sfor35min).TheGC
ovenhadtwocapillarycolumnsintandem(BP-1,10m,ID0.53mm, 2.65m filmthickness respectively) from Agilent Technologies
(Avondale,PH,USA).Thedetectorandinjectortemperatureswere at300and220◦C,respectively.Heliumwasusedascarriergasat
a constant flowrate of7ml/min. For FPDoperationthe
hydro-gen flow was setat 90ml/min and theair one at 115ml/min.
Heliumwasusedasthedetectormakeupgasat30ml/min.The
temperatureprogramoftheGCovenwas:initialT50◦C(holdfor
1min),increasedto170◦Cat16◦C/min,rampedto220◦Catthe
rateof6◦C/min(holdfor1min),increasedto240◦Cattherateof
4◦C/min,finallyto280◦Cattherateof5◦C/min(holdfor10min)
andreturnedtoinitialconditionsin5min.Totalruntime40.8min. Theinjectionvolumewas2l.Thesoftwareforthecontrolofthe
GC–FPDwasChromCard,ThermoFinnigan(Rodano,Milan,Italy). Residueconfirmationinrealsampleanalysiswereperformedin aTrace2000GCequippedwithaThermoQuestautosampler(model AS2000),asplit/splitlessinjectorconnectedwiththeGCQplus ion-trapmassspectrometer(Thermoquest,Austin,TX,USA),operating ineitherMSnorSIMmodes,injecting2
lofthetestedsolutions.
TheoperationconditionsoftheGCQPlusMS systemwere: the
injectorin splitlessmodeunderisothermalconditionsat220◦C
andthesplitvalvewasopened1minaftertheinjection.Gas chro-matographywascarriedoutonDB-5MS(J&WScientific)0.25m,
30m×0.25mmwitha1m×0.25mmi.d.guardcolumnof
deacti-vatedfusedsilica(Alltech,Augsburg,Germany).Oventemperature gradientwasprogrammedasfollows:theinitialtemperaturewas 50◦Cfor1min,andincreasedto120◦Cattherateof22.5◦C/min,
rampedto250◦Cat3◦C/minfor1minandthenincreasedto285◦C
attherateof15◦C/minwhichwasheldfor10minandreturned
totheinitialconditionsin5min.Heliumwasthecarriergasata flowrateof1ml/min.TheMSsystemwasoperatedintheelectron impactionizationwithpositivepolarityionmode. Theemission currentwas250mA,themultipliervoltagewas1700Vandafull
scanrangewassetto50–500amuwithmaximumiontime25ms,
10microscansandAGCtargetvalueof50.Thetransferlineand themanifoldtemperatureweresetat285and220◦C,respectively.
AnalyteswereidentifiedbycomparingtheirEImassspectrawith home-madelibraries.
Extractionprocedures
MAE
Drymateleaves(5g)wereweighedandputintothe
vesselandshakedvigorouslybyhandfor30s.Setsof12vessels weremicrowaveextractedaccordingtothefollowingoperational
parameters;magnetronpower800W,maximumpressure100psi,
heatedto80◦Cin10minandmaintainedfor15min.
Afterremovingthevesselsfromthemicrowaveoven,theywere cooledatroomtemperature.Theextractfromeachvesselwas fil-teredundervacuumandrinsedwith15mlMeCN.A15mlaliquot wastransferredtoatubecontaining1mltolueneandevaporated untildrynessunderN2stream.Samplecleanupconsistedintwo stepsfollowingamodificationofthemethoddescribedin2003by Haibetal.First,thedryextractwasre-dissolvedin1mlofMeCN andloadedintoa690mgsilicacartridgefollowedbytheaddition of0.5mloftoluene.Thetargetcompoundswereelutedwith3ml ofanacetone–toluene(8:2)mixture.The3mleluatewasloaded intoa500mgENVI-carbcartridgeandelutedwith3mlofacetone. Eachcartridgewaspre-conditionedwith5mlofacetone.Thefinal eluatewascollected,thesolventevaporatedandtheresiduewas dissolvedin200lofEtOAcforGC–FPDanalysis.
QuEChERS
The employed procedure was a modification of the citrate
bufferedQuEChERSmethodCEN15662(www.cen.eu),(Payáetal.,
2007;Anastassiadesetal.,2010).Arepresentative2gsamplewas
weighed in a 50ml PTFE centrifugation tube. Afterwards, 10g
ofchopped ice and 10ml of MeCNwere addedinto each tube
(Haywardetal.,2013;Rajskietal.,2013).Then4gofMgSO4,1gof
NaCl,0.5gofsodiumcitratedibasicsesquihydrateand1gofsodium citratetribasicdihydratewereadded.Thetubewashandshaken for4minandcentrifuged,10minat3000×g.Forthecleanupstep,
a6mlaliquotoftheextractwastransferredtoa15mlPTFE cen-trifugationtubecontaining855mgofMgSO4,150mgofPSAand 45mgofGCB.Thistubewasshakenfor20susingavortexand cen-trifugedfor10minat3000×g.Afterthat40lof5%formicacidin
MeCNwereaddedto4mlofextractanda1mlaliquotwas trans-ferredtoa5mlconictubeandevaporatedundernitrogenstream untildryness.Finally,theextractwasdissolvedin200lofEtOAc
forGC–FPDanalysis.
Resultsanddiscussion
Extractionandcleanupoptimization
The analysisof pesticide residues using microwave assisted extractionsystems requirethe optimizationof different opera-tionalparameterssuchasmagnetronpower,temperature,pressure andextractiontime.Theoptimumconditionsfortheextractionof pesticidesbyMAEindifferentmatriceswereselectedtakinginto considerationpreviousreports(Nielletal.,2011;Vryzasetal.,2002,
2007;Papadakisetal.,2006;VryzasandPapadopoulou-Mourkidou,
2002). QuEChERSand MAEprotocolsyielded highly pigmented
extractsandGCBwasused intheclean upsteptoremove the
co-extractedchlorophyll.However,theamountofGCBusedwas
abalancebetweentherecoveriesofthestudiedpesticidesandthe pigmentremoval.IntheMAEprotocol,anENVICARBcartridgewas used,accordingtothemethodproposedbyHaywardetal.forherbs,
whereasQuEChERSusedGCBandPSAinadispersivemode.
Nev-ertheless,PSAwasnotemployedinMAEmethod,aspolyphenols
andshikimicacidanalogs suchaschlorogenicacidpresentinI. paraguaiensiscouldbeanalyteprotectantsforthemostlabile pes-ticidesbyinteractingwiththesilvnolfreeOHintheglasslinerasit hasbeenestablishedintheliterature(Anastassiadesetal.,2003).
Methodsperformanceandvalidation
Allvalidationprocedureswereperformedusingacommercial
yerba mate sample labeled as organic, which was previously
80
70
60
50
40
30
20
10
0 14
22
> 50% 25-50% < 25%
ME QuECHERS ME MAE
% pesticides 19 19
67 59
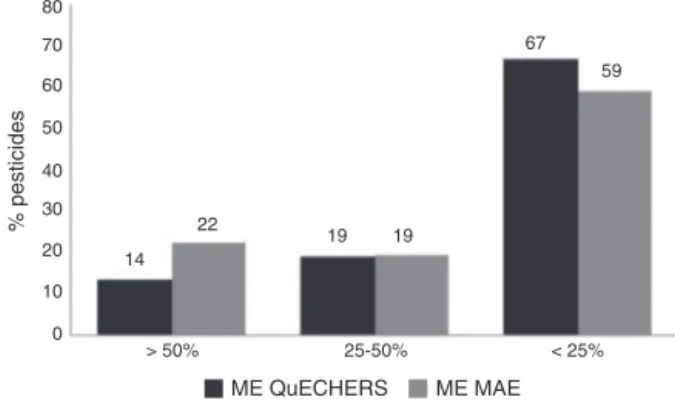
Fig.1.CalculatedmatrixeffectsofMAEandQuEChERSmethod.Matrixeffect (%)=(1−(slopematrix/slopesolvent))×100.
analyzedinordertodeterminethepre-existentpesticideresidues content.
Themethodefficiency,expressedasrecoveryratesandrelative standarddeviation(%RSD)ofthetestedpesticides,wasdetermined attwofortificationslevels:0.2and0.5mg/kginspikedsamplesof yerbamate,asitisshowninTable1.
Among the 42 pesticides included in the analytical method
phorate,fenthion,terbufos,fenamiphos,andmetamidofosexhibit recoverieslowerthan50%forbothmethodsandcannotbe
deter-minedaccordingtoDG-SANCOguidelines(EuropeanCommission
DG-SANCO,2014).Theremaininganalytespresenteddifferences
intherecoveryresultsforbothmethods.ParticularlywithMAE extraction,fensulfothionwasnotdetectedatanyfortificationlevel,
whiledichlorvos,phosphamidonanddimefoxpresented
recover-iesbetween19and63%at0.2mg/kg.QuEChERSmethodpresented lowrecoveriesforomethoateat0.2mg/kg,prothiofosatboth lev-elsandchlorpyrifospresentedrecoveriesof65and59%at0.2and 0.5mg/kgrespectively.
Theselowrecoveriescouldbeduetothepossiblevolatilization ordegradationduringGCdetermination(Ingelseetal.,2001)ordue tothelossesduringtheconcentrationprocess.Itwasobservedthat mostofthepesticidesshowinglowrecoveriesarevolatileandhave thesmallestretentiontimes(Table1).ConcerningtheQuEChERS methodmostofthepesticideswithlowrecoverieselutedinthe middleofthechromatogramandaftercaffeine.
AsitisshowninFig.1,QuEChERSmethodshowedlowermatrix
effectthan MAE.Signalenhancement wasobserved for 41 and
33%ofthestudiedpesticidesinMAEandQuEChERS,respectively.
Particularly mevinphos showed 75% of signal enhancement in
QuEChERSmethod,thiscouldleadtooverquantification,aspointed
out by theDG-SANCO guidelines, explaining thehighrecovery
observed.
Matrix-matchedcalibration curves werelinear in the range 0.05–4g/mlwithcorrelationcoefficients(r2)higherthan0.99in
mostcases.Onlydichlorvospresentedlinearityproblemsin QuECh-ERSandthiscouldbeattributedtoitshighvolatilityandthermal lability.TheseproblemswerenotobservedinMAE,supportingthe hypothesisoftheanalyteprotectanteffectofmatepolyphenols.
Thelimitsofdetection(LOD),rangedfrom0.004to1mg/kg. TheLOQ,determinedasestablishedinDG-SANCOguidelinesisthe lowestconcentrationoftheanalytethathasbeenvalidatedwith acceptableaccuracybyapplyingthecompleteanalyticalmethod, rangedfrom0.1to0.2mg/kgformostoftheevaluatedpesticides. However,consideringtheLOQastheLOD×10,28/33pesticides
presenteda LOQbelow0.2mg/kginMAEand 11/27in
QuECh-ERS. Somepesticidessuch asphenthoate, prothiofos,parathion
ethyl,omethoate,dimefoxandchlorpyrifosinQuEChERSmethod
Table1
(%)RecoveryratesandrespectiveRSDobtainedforMAEandQuEChERSmethodat0.2and0.5mg/kgspikinglevels(pesticideswithacceptablerecoveriestooneatleastof thetestedmethodswereonlyincluded).
Pesticide commonname
Stock mix
RT (min)
Spikinglevel (mg/kg)
MAE QuEChERS
Recovery(%) RSD(%) Recovery(%) RSD(%)
Acephate I 11.59 0.20.5 8492 43 7077 49
Bromophos
methyl III 21.98
0.2 97 11 75 11
0.5 93 6 65 11
Cadusafos III 15.85 0.20.5 8480 114 9991 49
Chlorfenvinphos I 22.68 0.20.5 8994 53 9374 124
Chlorpyrifos III 21.19 0.2 86 11 65 4
0.5 82 5 59 13
Chlorpyrifos
methyl II 19.29
0.2 90 6 76 8
0.5 89 10 73 6
Diazinon I 17.48 0.20.5 9188 14 7776 154
Dichlorvos II 9.55 0.2 63 16 99 4
0.5 67 15 109 10
Dimefox II 7.38 0.2 50 15 85 6
0.5 53 14 113 14
Dimethoate II 16.13 0.20.5 112109 63 10796 148
Disulfoton
sulfoxide III 10.50
0.2 91 9 117 12
0.5 90 4 117 9
Disulfotonsulfone I 23.47 0.2 105 2 119 5
0.5 110 1 95 21
Ethion III 26.64 0.2 103 9 76 3
0.5 101 4 65 16
Ethoprophos II 14.91 0.20.5 100101 64 7890 69
Fenchlorphos II 19.96 0.2 95 5 70 4
0.5 91 9 67 6
Fenitrothion III 19.99 0.2 118 11 99 5
0.5 111 3 91 13
Fonofos III 17.26 0.20.5 8680 124 8576 114
Fensulfothion II 26.67 0.2 ND ND 82 17
0.5 ND ND 89 9
Heptenophos III 13.78 0.2 90 12 119 4
0.5 85 5 116 7
Malathion II 20.52 0.20.5 10499 38 8088 39
Mecarbam III 22.49 0.20.5 10097 103 10293 124
Methidathion II 23.28 0.2 107 7 102 10
0.5 103 9 98 7
Mevinphos III 11.69 0.2 88 13 131 3
0.5 83 6 134 4
Omethoate II 13.98 0.20.5 9379 1216 4981 1825
Parathion
ethyl II 21.16
0.2 103 4 61 5
0.5 96 9 74 9
Parathionmethyl III 19.14 0.2 118 12 116 3
0.5 117 1 100 11
Phenthoate II 22.76 0.20.5 109101 38 6676 38
Phosphamidon I 6.24 0.20.5 1922 1210 8780 78
Pirimiphosmethyl I 20.30 0.2 100 18 74 6
0.5 91 4 64 14
Profenofos III 24.74 0.20.5 118113 134 8877 147
Prothiofos II 24.83 0.20.5 9994 59 4350 75
Quinalphos III 22.70 0.2 89 11 93 5
0.5 86 6 85 12
Terbufossulfone II 22.30 0.2 106 3 88 2
0.5 101 8 86 11
Thionazin II 14.41 0.20.5 9796 74 7894 106
Tolclofosmethyl I 19.50 0.2 88 2 75 4
0.5 89 3 64 12
Triazophos I 26.73 0.2 98 6 114 3
0.5 104 3 80 14
Table2
Limitsofdetection(LOD)andlimitsofquantification(LOQ)inmg/kginGC/FPD.
Pesticidecommonname MAEmg/kg QuEChERSmg/kg MRL(EU)mg/kg
LOD LOD×10/LOQ LOD (LOD×10/LOQ
1.Acephate 0.01 0.1/0.2 0.05 0.5/0.2 0.05
2.Bromophosethyl 0.004 0.04/0.2 0.05 0.5/0.2 0.1
3.Cadusafos 0.004 0.04/0.2 0.01 0.1/0.2 0.01
4.Chlorfenvinphos 0.01 0.1/0.2 0.05 0.5/0.2 0.05
5.Chlorpyrifos 0.004 0.04/0.2 0.1 1.0/1.0 0.5
6.Chlorpyrifosmethyl 0.004 0.04/0.2 0.05 0.5/0.2 0.1
7.Diazinon 0.004 0.04/0.2 0.01 0.1/0.2 0.05
8.Dichlorvos 0.01 0.1/0.5 0.05 0.5/0.2 0.02
9.Dimefox 0.05 0.5/1.0 0.05 0.5/0.2 0.01
10.Dimethoate 0.05 0.5/0.2 0.05 0.5/0.2 0.1
11.Disulfotonsulfoxide 0.01 0.1/0.2 0.05 0.5/0.2
0.05
12.Disulfotonsulfone 0.004 0.04/0.2 0.01 0.1/0.2
13.Ethion 0.004 0.04/0.2 0.05 0.5/0.2 0.05
14.Ethoprophos 0.004 0.04/0.2 0.01 0.1/0.2 0.02
15.Fenchlorphos 0.05 0.5/0.2 0.05 0.5/0.2 0.1
16.Fenitrothion 0.01 0.1/0.2 0.05 0.5/0.2 0.05
17.Fonofos 0.004 0.04/0.2 0.01 0.1/0.2 0.01
18.Fensulfothion 1.0 1.0/1.0 0.05 0.5/0.2 0.01
19.Heptenophos 0.004 0.04/0.2 0.01 0.1/0.2 0.01
20.Malathion 0.05 0.5/0.2 0.05 0.5/0.2 0.02
21.Mecarbam 0.01 0.1/0.2 0.05 0.5/0.2 0.1
22.Methidathion 0.004 0.04/0.2 0.05 0.5/0.2 0.1
23.Mevinphos 0.004 0.04/0.2 0.01 0.1/0.2 0.02
24.Omethoate 0.01 0.1/0.2 0.05 0.5/0.5 0.05
25.Parathionethyl 0.01 0.1/0.2 0.05 0.5/0.5 0.1
26.Parathionmethyl 0.004 0.04/0.2 0.01 0.1/0.2 0.05
27.Phenthoate 0.01 0.1/0.2 0.05 0.5/0.5 0.01
28.Phosphamidon 0.01 0.1/1.0 0.05 0.5/0.2 0.02
29.Pirimiphosmethyl 0.004 0.04/0.2 0.01 0.1/0.2 0.3
30.Profenofos 0.01 0.1/0.2 0.05 0.5/0.2 0.1
31.Prothiofos 0.01 0.1/0.2 0.05 0.5/1.0 0.01
32.Quinalphos 0.004 0.04/0.2 0.01 0.1/0.2 0.1
33.Terbufossulfone 0.004 0.04/0.2 0.01 0.1/0.2 0.01
34.Thionazin 0.004 0.04/0.2 0.01 0.1/0.2 0.01
35.Tolclofosmethyl 0.05 0.5/0.2 0.05 0.5/0.2 0.1
36.Triazophos 0.004 0.04/0.2 0.05 0.5/0.2 0.02
37.Trichlorfon 0.004 0.04/0.2 0.01 0.1/0.2 0.05
methodshowedLOQhigherthan0.2mg/kg.astheycouldnotbe validatedwithacceptableaccuracyatthislevel(Table2).
Chromatographicanalysis
Twomegaborecolumnsintandemwereusedinordertoachieve
adequatechromatographicseparation. Megaborecolumns
(typi-cally10m×0.53mm)areadvantageouscomparedtonarrow-or
micro-borecolumnswhenextractsof“difficult”matriceshavetobe analyzedsincemegaborecolumnscanprovidehighloadabilityas filmsupto5m(Cajkaetal.,2008;Ravindraetal.,2008.).Theuseof
twomegaborecolumnsintandem(20m×0.53mm×2.65m)can
alsoimprovethechromatographicseparationofpesticideswith similarproperties,akeypointwhentheGCisnotconnectedwith aMSdetector(MastovskaandLehotay,2003).Therefore,the selec-tionofacolumnwithhighinternaldiameter(0.53mm)andfilm thickness(2.65m)ensurebetterperformancein sampleswith
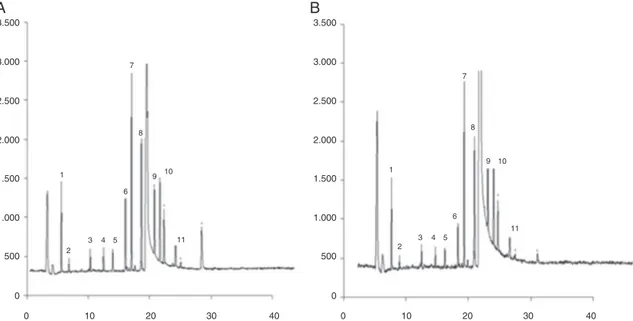
highmatrixeffect.Alongoventemperaturegradientwasselected (runtime40.8min)toimprovethechromatographicresolutionof theanalyteswhicharedifficulttoresolveundertypicalGC con-ditions.TheOPpesticidesincludedintheanalyticalmethodwere separatedinthreestocksolutionsbasedontheretentiontimeof eachanalyte(Table1).Separationoftargetcompoundswas per-formedinordertoavoidco-elutionofsomepesticides.Fig.2shows thechromatogramobtainedfortheanalysisoffortifiedyerbamate
sampleswithMixIat0.1mg/kgwithbothMAEandQuEChERS
methodsbyGC–FPD.
Asitispresentedinthechromatograms(Fig.2),thereisapeak withretentiontimearound20mincorrespondingtocaffeine.The
cleanupofbothmethodsisnotenoughtoremoveallthecaffeine, althoughMAEcleanupismoreefficientthanQuEChERS.
Realsampleanalysis
Inordertochecktheperformanceof themethodnine
com-mercialsampleswereanalyzed.Thesampleswereextractedusing bothvalidatedmethodsandanalyzedbyGC/FPDandthepositive findingswereconfirmedbyGC/MS.
Acephate, ethoprophos, chlorpyrifos, and cadusafos were
detected in commercial samples and their concentrations are
showninTable3.However,onlychlorpyrifosshowed concentra-tionsabovetheLOQofMAEmethodinfivesamplesandbelowthe correspondingMRL(EuropeanCommission,2005,2014).
MAEandQuEChERScomparison
TheanalyticalresultsofrealsamplesshowninTable3indicate that,undertheexperimentalconditionsemployedinthepresent
communication, MAE provides betterextractability of incurred
residuespresentinrealsamplesasitdetectsnotonlymore pes-ticidesbutalsotheresidueconcentrationsfoundarehigherthan QuEChERS.
Thereasonoftheseresultscouldbebasedintheefficiencyof microwaveenergy,whichishigherthanmanualagitationforthe extractionoftheresiduesfromthematrix.
Concerning matrix effect MAE presented more compounds
3.500
3.000
2.500
2.000
1.500
1.000
500
0
0 10
1
2 3 4 5
6 7
8
9 10
11
A
B
20 30 40
3.500
3.000
2.500
2.000
1.500
1.000
500
0
0 10
1
2 3 4 5
6 7
8
9 10
11
20 30 40
Fig.2. ChromatogramsoffortifiedmatesampleswithMixIat0.1mg/kgbyGC–FPD.MAE(A)andQuEChERS(B)methods.1:trichlorfon;2:phosphamidon;3:acephate;4: phorate;5:diazinon;6:tolclofosmethyl;7:pirimiphosmethyl;8:fenthion;9:chlorfenvinphos;10:disulfutonsulfone;11:triazophos.
Table3
Pesticides(mg/kg)detectedbyGC–FPDandconfirmedbyGC–MSinrealsamples.ND:notdetected.
Realsample Acephate Ethoprophos Chlorpyrifos Cadusafos
MAE/QuEChERS MAE/QuEChERS MAE/QuEChERS MAE/QuEChERS
1 <LOQ/ND <LOQ/ND ND ND
2 ND ND 0.3/<LOQ ND
3 ND ND <LOQ/ND ND
4 ND ND ND ND
5 ND ND <LOQ/ND ND
6 ND ND 0.2/<LOQ ND
7 ND ND 0.4/<LOQ ND
8 ND ND 0.2/ND <LOQ/ND
9 ND ND 0.2/ND <LOQ/ND
moreeffectiveavoidingtheextractionofcaffeine,whichisthemain detectedinterference.
Ingeneral,MAEmethodensuredlowertosimilarLODforall pes-ticidesexceptforfensulfothion,comparedwithQuEChERSmethod, whiletheLOQ(lowestvalidatedlevel)for28 pesticidesinboth methodsweresimilar.IfLOQarecalculatedasLOD×10,13 pesti-cidescouldbeassessedforMRLcompliancewithMAEmethodand threepesticideswithQuEChERS.
Comparingtheaccuracyandprecisionofbothmethods,MAE presentedbetterperformancethanQuEChERS,sincetherecoveries of33pesticideswerewithintherange70–118%withRSDsfrom1 to18%.QuEChERSmethodpresentedrecoveryratesbetween70 and120%andRSDsintherange3–21%for27pesticides,atthe lowestspikinglevel.Someoftheobtainedresultsinthisstudywith QuEChERSmethodweresimilartothosereportedbyLozanoetal.
(2012),indifferenttypesofteausingGC-QqQ/MS.
QuEChERSmethodologyissimple,cheap,practicallyno glass-wareis needed,and it ismore environmentally friendlyasthe solventconsumptionislowerthanMAE.
MAEpresentedgoodperformance,itallowsthesimultaneous extractionof10samples,buttheequipmentrequiredisnotoften availableinthelaboratories.
Thepresentstudydemonstratedthatalthoughbothmethods
aresuitablefortheanalysisofpesticideresiduesinyerbamate,MAE presentedabetterperformanceundertheexperimentalconditions tested.
Yerbamateisconsumeddailybyalmost50millionpeoplebut therearefewdataontheliteratureconcerningthepersistenceof pesticideresiduesintheprocessedleaves.Thisworkmighthelp
togathertheinformationneededtoperformstudiesonpesticide residueexposureofthepopulationduetoyerbamateintake.
Authors’contributions
JG,SNandLPperformedthelaboratorywork,dataand chro-matographicanalysis.HHandZVranthefirsttrialexperiments withMAE.ZVanalyzedtherealsamplesintheGC–(ITD)-MS,LP,ZV andSNdraftedthepaper.VC,LPandHHgavetheworksconceptual frame,participated intheresultsdiscussionandthemanuscript finalwriting.ZVandEPMsupervisedthelaboratoryworkandZV contributedtocriticalreadingofthemanuscript.
Conflictofinterest
Theauthorshavenoconflictofinteresttodeclare.
Acknowledgment
TheauthorsgratefullyacknowledgetheEuropeanCommission
(Alfa II Programme B-Project EUROLANTRAP, No.
AML/B7-311/97/0666/II0461-FA-FCD-FI).
References
Anastassiades,M.,Hepperle,J.,Roux,D.,Sigalov,I.,Mack,D.,2010.Extractability ofincurredresiduesusingQuEChERS.EuropeanPesticideResidueWorkshop. EPRW,Strasbourg,France.
Andrade,F.,deAlbuquerque,C.A.C.,Maraschin,M.,daSilva,E.L.,2012.Safety assess-mentofyerbamate(llexparaquariensis)driedextract:resultsofacuteand90 dayssubchronictoxicitystudiesinratsandrabbits.FoodChem.Toxicol.50, 328–334.
Attallah,E.R.,Barakat,D.A.,Maatook,G.R.,Badawy,H.A.,2012.Validationofaquick andeasy(QuEChERS)methodforthedeterminationofpesticidesresiduein driedherbs.J.FoodAgric.Environ.10,755–762.
Cajka,T.,Sandy,C.,Bachanova,V.,Drabova,L.,Kalachova,K.,Pulkrabova,J.,Hajslova, J.,2012.Streamliningsamplepreparationandgaschromatography–tandem massspectrometryanalysisofmultiplepesticideresiduesintea.Anal.Chim. Acta743,51–60.
Cajka,T.,Hajslova,J.,Lacina,O.,Mastovska,K.,Lehotay,S.J.,2008.Rapidanalysisof multiplepesticideresiduesinfruit-basedbabyfoodusingprogrammed temper-aturevaporiserinjection–low-pressuregaschromatography–high-resolution time-of-flightmassspectrometry.J.Chromatogr.A1186,281–294.
Chen,G.,Cao,P.,Liu,R.,2011.Amulti-residuemethodforfastdeterminationof pesticideinteabyultraperformanceliquidchromatography–electrospray tan-demmassspectrometrycombinedwithmodifiedQuEChERSsamplepreparation procedure.FoodChem.125,1406–1411.
Chen,Y.,Al-Taher,F.,Juskelis,R.,Wong,J.W.,Zhang,K.,Hayward,D.G., Zweigen-baum,J.,Stevens,J.,Cappozzo,J.,2012a.Multiresiduepesticideanalysisofdried botanicaldietarysupplementsusinganautomateddispersiveSPEcleanupfor QuEChERSandhigh-performanceliquidchromatography–tandemmass spec-trometry.J.Agric.FoodChem.60,9991–9999.
Chen,L.,Songa,F.,Liua,Z.,Zhenga,Z.,Xinga,J.,Liua,S.,2012b.Multi-residuemethod forfastdeterminationofpesticideresiduesinplantsusedintraditionalChinese medicinebyultra-high-performanceliquidchromatographycoupledtotandem massspectrometry.J.Chromatogr.A1225,132–140.
Cho,S.,AbdEl-Aty,A.M.,Choi,J.,Jeong,Y.,Shin,H.,Chang,B.,Lee,C.,Shim,J.,2008.
Effectivenessofpressurizedliquidextractionandsolventextractionforthe simultaneousquantificationof14pesticideresiduesinGreenteausingGC.J. Sep.Sci.31,1750–1760.
EuropeanCommission,2005.Regulation(EC)No.396/2005oftheEuropean Par-liamentandoftheCouncilof23February2005onmaximunresiduelevelsof pesticidesinoronfoodandfeedofplantandanimaloriginandamendingCouncil Directive91/414/EEC.Off.J.Eur.Union,1–16.
EuropeanCommission DG-SANCO,2014. Method validationand quality con-trolproceduresforpesticide residueanalysisinfoodandfeed.Document SANCO/12571/2013,1January.
EU-MRLs Database. http://ec.europa.eu/sancopesticides/public/?event= commodity.resultat(accessedJune2014).
Filip,R.,Lopez,P.,Giberti,G.,Coussio,J.,Ferraro,G.,2001.Phenoliccompoundsin sevenSouthAmericanIlexSpecies.Fitoterapia72,774–778.
Haib,J.,Hofer,I.,Renaud,J.M.,2003.Analysisofmultiplepesticideresiduesintobacco usingpressurizedliquidextraction,automatedsolid-phaseextractioncleanup andgaschromatography–tandemmassspectrometry.J.Chromatogr.A1020, 173–187.
Hayward,D.,Wong,J.W.,Shi,F.,Zhang,K.,Lee,N.S.,DiBenedetto,L.,Hengel,M., 2013.Multiresiduepesticideanalysisofbotanicaldietarysupplementsusing salt-outacetonitrileextraction,solid-phaseextractioncleanupcolumn,and gaschromatography–triplequadrupolemassspectrometry.Anal.Chem.85, 4686–4693.
Heck,C.I.,deMejia,E.G.,2007.Yerbamatetea(Ilexparaguariensis):acomprehensive reviewonchemistry,health,implications,andtechnologicalconsiderations.J. FoodSci.72,R138–R148.
Huang,Z.,Li,Y.,Chen,B.,Yao,S.,2007.Simultaneousdeterminationof102 pes-ticideresiduesinChineseteasbygaschromatography–massspectrometry.J. Chromatogr.B853,154–162.
Huang,Z.,Zhang,Y.,Wang,L.,Ding,L.,Wang,M.,Yan,H.,Li,Y.,Zhu,S.,2009. Simul-taneousdeterminationof103pesticideresiduesinteasamplesbyLC–MS/MS. J.Sep.Sci.32,1294–1301.
Ingelse,A.B.,vanDam,C.J.R.,Vreeken,J.R.,Mol,G.J.H.,2001.Determinationofpolar organophosphoruspesticidesinaqueoussamplesbydirectinjectionusingliquid chromatography–tandemmassspectrometry.J.Chromatogr.A918,67–78.
Jacques,R.A.,dosSantosFreitas,L.,FloresPeres,V.,Dariva,C.,Oliveira,J.V.,Camarão, E.B.,2006.Chemicalcompositionofmatetealeaves(Ilexparaguariensis):astudy ofextractionmethods.J.Sep.Sci.29,2780–2784.
Jacques,R.A.,Santos,J.G.,Dariva, C.,Oliveira, J.V.,Camarão,E.B.,2007.GC/MS characterizationofmatetealeavesextractsobtainedfromhigh-pressureCO2
extraction.J.Supercrit.Fluids40,354–359.
Kanrar,B.,Mandal,S.,Bhattacharyya,A.,2010.Validationanduncertainty analy-sisofamultiresiduemethodfor42pesticidesinmadetea,teainfusionand spentleavesusingethylacetateextractionandliquidchromatographymass spectrometry.J.Chromatogr.A1217,1926–1933.
Lozano, A., Rajski, Ł., Belmonte-Valles, N., Uclés, A., Uclés, S., Mezcua, M., Fernández-Alba,A.R.,2012.Pesticideanalysisinteasandchamomileby liq-uidchromatographyandgaschromatographytandemmassspectrometryusing amodifiedQuEChERSmethod:validationandpilotsurveyinrealsamples.J. Chromatogr.A1268,109–122.
Khan,Z.S.,Ghosh,R.K.,Girame,R.,Utture,S.C.,Gadgil,M.,Banerjee,K.,Reddy, D.D.,Johnson,N.,2014.Optimizationofasamplepreparationmethodfor mul-tiresidueanalysisofpesticidesintobaccobysingleandmulti-dimensionalgas chromatography–massspectrometry.J.Chromatogr.A1343,200–206.
Liu, D.,Min,S.,2012. Rapidanalysisof organochlorineandpyrethroid pesti-cides inteasamples by directlysuspended dropletmicroextractionusing a gas chromatography–electron capture detector. J. Chromatogr. A 1235, 166–173.
Mastovska, K., Lehotay, S.J., 2003. Practical approaches to fast gas chromatography–massspectrometry.J.Chromatogr.A1000,153–180.
Moinfar,S.,Hosseini,M.M.,2009.Developmentofdispersiveliquid–liquid microex-tractionmethodfortheanalysisoforganophosphoruspesticidesintea.J.Hazard. Mater.169,907–911.
Nguyen,T.D.,Lee,K.J.,Lee,M.H.,Lee,G.H.,2010.Amultiresiduemethodforthe determinationof234pesticidesinKoreanherbsusinggaschromatographymass spectrometry.Microchem.J95,43–49.
Niell,S.,Pareja,L.,González,G.,González,J.,Vryzas,Z.,Cesio,M.V., Papadopoulou-Mourkidou,E.,Heinzen,H.,2011.Simpledeterminationof40organophosphate pesticidesinrawwoolusingmicrowave-assistedextractionandGC–FPD anal-ysis.J.Agric.FoodChem.59,7601–7608.
Papadakis,E.,Vryzas,Z.,Papadopoulou-Mourkidou,E.,2006.Rapidmethodforthe determinationof16organochlorinepesticidesinsesameseedsby microwave-assisted extractionand analysisof extractsby gas chromatography–mass spectrometry.J.Chromatogr.A1127,6–11.
Payá,P.,Anastassiades,M.,Mack,D.,Sigalova,I.,Tasdelen,B.,Oliva,J.,Barba,A.,2007.
AnalysisofpesticideresiduesusingtheQuickEasyCheapEffectiveRuggedand Safe(QuEChERS)pesticidemultiresiduemethodincombinationwithgasand liquidchromatographyandtandemmassspectrometricdetection.Anal.Bioanal. Chem.389,1697–1714.
PérezParada,A.,González,J.,Pareja,L.,GeisAsteggiante,L.,Colazzo,M.,Niell,S., Besil,N.,González,G.,Cesio,V.,Heinzen,H.,2010.Transferofpesticidestothe brewduringmatedrinkingprocessandtheirrelationshipwithphysicochemical properties.J.Environ.Sci.HealthB45,1–8.
Rajski,L.,Lozano,A.,Belmonte-Valles,N.,Uclés,A.,Uclés,S.,Mezcua,M., Fernandez-Alba,A.R.,2013.Comparisonofthreemultiresiduemethodstoanalysepesticides ingreenteawithliquidandgaschromatography/tandemmassspectrometry. Analyst138,921–931.
Ravindra,K.,Dirtu,A.C.,Covaci,A.,2008.Low-pressuregaschromatography:recent trendsanddevelopments.TrendsAnal.Chem.27,291–303.
Sadowska-Rociek,A.,Surma,M.,Cie´slik,E.,2013.ApplicationofQuEChERSmethod forsimultaneousdeterminationofpesticideresiduesandPAHsinfreshherbs. Bull.Environ.Contam.Toxicol.90,508–513.
Vázquez,A.,Moyna,P.,1986.Studiesonmatedrinking.J.Ethnopharmacol.18, 267–272.
Vryzas,Z.,Papadakis,E.N.,Papadopoulou-Mourkidou,E.,2002.Microwave-assisted extraction(MAE)-acidhydrolysis ofdithiocarbamatesfor traceanalysisin tobaccoandpeaches.J.Agric.FoodChem.50,2220–2226.
Vryzas, Z., Papadopoulou-Mourkidou, E., 2002. Determination of triazine and chloroacetanilideherbicidesinsoilsbymicrowave-assistedextraction(MAE) coupledtogaschromatographicanalysiswitheitherGC–NPDorGC–MS.J.Agric. FoodChem.50,5026–5033.
Vryzas, Z.,Tsaboula, A., Papadopoulou-Mourkidou, E., 2007.Determination of alachlor,metolachlor,andtheir acidicmetabolites insoils by microwave-assistedextraction(MAE)combinedwithsolidphaseextraction(SPE)coupled withGC–MSandHPLC–UVanalysis.J.Sep.Sci.30,2529–2538,www.cen.eu