* e-mail: [email protected]
colhida na Guiné-Bissau, conduziu ao isolamento de uma biblioteca de 41 compostos alquil- e alquenil-fenólicos, cujas estruturas foram elucidadas por RMN 1H e 13C, e CGEM. A localização das insaturações nas cadeias laterais dos alquenil-fenóis foi determinada por CGEM dos respectivos derivados metiltiolados.
Bioassay-guided fractionation of the ethanolic root extract of Ozoroa insignis, collected in Guinea-Bissau, led to the isolation of a 41-member library of alkyl and alkenylphenols, whose structures were determined by 1H and 13C NMR, and GCMS. Determination of double-bond positions in the side chains of alkenylphenols were established by methylthiolation-GCMS.
Keywords: Ozoroa insignis, Anacardiaceae, cardanols, anacardic acid methyl esters, GCMS
Introduction
Ozoroa insignis Del. (Heeria insignis Del.) (Anacardiaceae) is a shrub or tree to 6.5 m high, of the soudanian savanna from Senegal to Niger and Nigeria, and across Africa to Ethiopia, Zaire and E. Africa.1 This species
has a wide range of healing properties, namely, in the treatment of diarrhea and venereal diseases,2 tapeworm and
hookworm,3 schistosomiasis,4,5 and kidney trouble.6 The
anthelmintic effect of root bark and leaves of O. insignis was evidenced in vitro against the worms Schistosoma mansoni and Hymenolepis diminuta,5 whereas a bark extract
displayed cytotoxic activity against Hep-G2, MDA-MB-231, and 5637 human cancer cell lines.2 Stembark and
stemwood were also investigated for in vitro topoisomerase inhibition.7 In Guinea-Bissau, the infusion of roots of O.
insignis is taken by women after childbirth to increase lactation.8 Although other traditional uses of the roots of
O. insignis have been reported,1 so far, only the leaves, flowers, twigs, and bark have been submitted to a limited phytochemical investigation that led to the isolation of essential oils, anacardic acid, and ginkgolic acid.2,6,9
In a previous antitumor screening of medicinal plants from Guinea-Bissau,8 we have observed that an ethanolic
root extract of O. insignis showed moderate activity in KB, A 549 and MDA-MB cell lines, with IC50 values of 30.5, 22.0 and 15.5 µg mL-1, respectively. In the present
paper, we describe the isolation and structural deter-mination of a series of alkyl and alkenylphenols from non-polar fractions of the ethanolic extract, whose activity was monitored by the brine shrimp assay.
Results and Discussion
The ethanol extract of O. insignis was fractionated according to Scheme 1, and the chromatographic fractions monitored by TLC and the brine shrimp assay.10,111H and 13C NMR, and GLC analysis indicated the presence of five
mixtures (I-V) of long chain alkyl and alkenylphenols (Figure 1), which were further characterized by GCMS of their α,β-bis-methylthiolated derivatives. The substitution pattern of the aromatic ring was assigned on the basis of the NMR data (see experimental), including COSY, DEPT, HMQC, HMBC and NOESY spectra, which were similar to those reported for cardanols and anacardic acids.6,12-17
GCMS of mixture I showed peaks of [M]+ at m/z 232,
276, 304, and 332, three peaks with the same [M]+ at m/z
302, two peaks of [M]+ at m/z 330, and one peak of [M]+ at
cardanols (6, 16-18, 21, 22, and 29) with monounsaturated side chains differing in the position of the double bond. In all cases, the mass spectra displayed a base peak at m/z 108 due to benzylic cleavage of alkyl 3-hydroxy-benzene.15 Location of the double bond in the alkenyl side
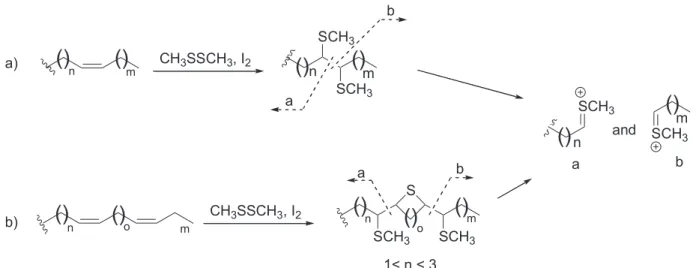
chain was deduced from mass spectral analysis of the corresponding α,β-bis-methylthiolated derivatives, which provide readily recognizable fragments on electron bombardment (Table 1; Figure 2a).18,19 The Z stereochemistry
of the double bond was assigned from the appearance of a low resolution multiplet in the 1H NMR spectrum at δ 5.40
(2H, m), and carbons at allylic positions at δ 27.2.17
The NMR profiles of mixtures II and III were identical to those of I. In mixture II, thirteen cardanols with monounsaturated side chains (7, 8, 10, 13-15, 19, 20, 23 -26, and 31) could be characterized from their GCMS and mass spectra fragmentation of the methylthiolated derivatives (Table 1; Figure 2a),18,19 whereas in mixture
III, in addition to five cardanols with saturated side chains (1-5), and six with monounsaturated side chains (9, 11,
12, 27, 28, 31) (Table 1), one compound with a diunsaturated side-chain (30) was identified. In this case, the derivatization reaction leads to a cyclic methylthiolated derivative, whose mass spectrum showed two fragments at m/z 279 (base peak) and m/z 201, indicative of the presence of two double bonds separated by two methylene groups (Figure 2b, o = 2).19
The NMR data of mixture IV, when compared to those of I-III, showed a different substitution pattern of the aromatic ring. Three coupled aromatic protons were observed at δ 6.72 (d, J 7.6 Hz), 6.85 (d, J 8.0 Hz) and δ 7.27 (t, J 8.0 Hz), and, in addition to the presence of the alkyl side chain and a phenolic hydroxyl group, a carboxyl methyl ester was assigned from a HMBC correlation of a carbonyl resonance at δC 172.0 with a methyl group at δ
3.96. Analysis of COSY, DEPT, HMQC, HMBC and NOESY spectra, and comparison with literature data allowed to confirm a basic structure of anacardic acid methyl esters for the constituents of mixture IV.6,13-17 Its
GCMS indicated the presence of three compounds (32,
35,and 36) with saturated side chains and three others (39-41) with one Z double bond.17 Mixture V was
comprised of cardanol 29, and anacardic acid methyl esters 32-38, which were identified as described above.
Anacardic acids are natural products isolated mainly from the Anacardiaceae family, but they are also present in fungi, marine organisms, and insects.20-22 These compounds
are known to possess a wide range of biological properties, such as antimicrobial, antitumoral, molluscicide, antifungal, insectide, regulator of gene expression, antioxidant, lipoxygenase and uncoupling activity of oxidative phosphorylation. They inhibit the generation of superoxide radicals by xanthine oxidase, and lower the levels of serum
cholesterol in rats.2,14,21-27 6-Oxa isosteres of anacardic acids
proved to be among the most potent inhibitors of the bacterial two-component regulatory systems KinA/SpoOF and NRII/NRI, reported to date.28 They also constitute some
of the active principles of Ginkgo biloba,29 with a recognized effect as inhibitors of prolyl endopeptidase and glycerol-3-phosphate dehydrogenase,30,31 antifeedant and cytotoxic
activities.17,32,33 In this plant they occur together with
cardanols (or ginkgols), whose antitumor activity has also been demonstrated.17
The pool of anacardic acid methyl esters and cardanols identified in the present work possess some unusual structural features. Their alkyl side chains varies from 13 to 25 carbons
13 (C20H32O) 288 382 279 (20) 103 (20) 23.45
14 (C21H34O) 302 396 237 (10) 159 (10) 35.92
15 (C21H34O) 302 396 279 (100) 117 (70) 34.31
16 (C21H34O) 302 396 293 (100) 103 (30) 37.70
17 (C21H34O) 302 396 307 (95) 89 (25) 38.84
18 (C21H34O) 302 396 335 (30) 61 (15) 39.05
19 (C22H36O) 316 410 279 (20) 131 (15) 29.67
20 (C23H38O) 330 424 279 (15) 145 (10) 31.70
21 (C23H38O) 330 424 307 (100) 117 (35) 43.85
22 (C23H38O) 330 424 321 (50) 103 (20) 44.64
23 (C24H40O) 344 438 279 (20) 159 (10) 35.92
24 (C25H42O) 358 452 279 (20) 173 (20) 37.92
25 (C26H44O) 372 466 279 (30) 187 (10) 42.16
26 (C27H46O) 386 480 279 (20) 201 (100) 35.60
27 (C31H54O) 442 536 293 (25) 243 (20) 38.52
28 (C31H54O) 442 536 307 (30) 229 (10) 45.11
29 (C31H54O) 442 536 391 (20) 145 (25) 36.92
30 (C31H52O) 440 566 279 (100) 201 (50) 37.64
31 (C31H54O) 442 536 405 (10) 131 (25) 22.85
32 (C23H38O3) 362 11.29
33 (C24H40O3) 376 16.20
34 (C25H42O3) 390 18.44
35 (C26H44O3) 404 24.07
36 (C30H52O3) 460 27.90
37 (C31H54O3) 474 24.10
38 (C32H56O3) 488 36.40
39 (C22H34O3) 346 440 281 (20) 159 (25) 14.20
40 (C26H42O3) 402 496 379 (30) 117 (10) 39.34
41 (C28H46O3) 430 524 281 (20) 243 (10) 30.09
in length, and monounsaturated side chains at C2, C5, C7, C10 to C14, C18 and C19 positions could be confirmed. A C25:2-Δ10,14 cardanol (30) was also identified. In alkylphenols
isolated from other Anacardiaceae the length do not exceeds 17 carbons, and when only one double bond is present, the position is most frequently Δ8.28 To our best knowledge,
compounds 6-19, 21-25, 27-31, and 35-41 are new naturally occurring substances. Moreover, this is the first reported case of isolation of anacardic acid methyl esters.
Experimental
General experimental procedures
NMR spectra were recorded on a Bruker ARX 400 NMR spectrometer (1H at 400 MHz; 13C at 100.61 MHz), using
CDCl3 as solvent and TMS as reference. FT-IR spectra were measured on a Perkin-Elmer 157G infrared spectrophoto-meter. GCMS were run on a Micromass GCTOF spectrometer at 70 eV, equipped with a J&W DB-5 column (30 m × 0.25 mm; 0.25 µm film thickness), helium flow rate at 4 mL min-1, splitless time 1 min, injector and detector
temperatures 280 °C. Oven temperature program was set at 120 °C to 240 °C at 10 °C min-1; 8 mn at 240 ºC; 240 °C
to 280 °C at 2 °C min-1, and 10 min at 280 °C. Celite,
Macherey-Nagel Si gel (0.04-0.063 mm/230-400 mesh), and Sephadex LH-20 were used for column chromatography (CC). TLC were performed on normal phase Si gel F254 plates (MN 818133), and visualized with Ce2SO4 and phosphomolybdic acid reagents.
Plant material
Ozoroa insignis Delile (Anacardiaceae), was collected in January 1994 at Contuboel, Guinea-Bissau, and
identified at the Herbarium of Botany Centre (LISC), where the voucher specimen nº 854 is deposited.
Extraction and fractionation
The air dried roots of O. insignis (1.65 kg) were powdered, and extracted in a Soxhlet apparatus with EtOH (5 L) at room temperature. Evaporation of the solvent under reduced pressure afforded an oily residue (83.3 g), that was submitted to fractionation according to Scheme 1.
Brine shrimp assay
The brine shrimp (Artemia salina) lethality assay was carried out according to a described method.10,11
Mixture I (1, 3, 5, 6, 16-18, 21, 22, 29)
IR (NaCl) νmax/cm-1: 2919, 2850, 2360, 1587, 1455,
1259, 1151, 1066. 1H NMR (CDCl
3, 400 MHz): δ 0.89 (t,
7.1, Me), 1.29 (bs), 1.65 (m, H-2'), 2.55 (t, 6.8, H-1'), 5.40 (m), 6.63 (d, 6.0, H-6), 6.66 (s, H-2), 6.75 (d, 7.2, H-4) and 7.13 (ddd, 7.6, 7.6, 2.5, H-5). 13C NMR CDCl
3,
100.61 MHz): δ 14.1 (Me), 22.7, 27.2, 29.3, 29.5, 29.6, 29.7, 30.4, 31.3, 32.0, 35.8 (C-1'), 112.5 (C-6), 115.3 (C-2), 121.0 (C-4), 129.4 (C-5), 129.9, 145.0 (C-3), 155.4 (C-1). For MS data see Table 1.
MixtureII (7, 8, 10, 13-15, 19, 20, 23-26, 31)
IR (NaCl) νmax/cm-1: 2919, 2850, 2360, 1587, 1455,
1259, 1151, 1066. 1H NMR (CDCl
3, 400 MHz): δ 0.89 (t,
7.1, Me), 1.29 (bs), 1.65 (m, H-2'), 2.55 (t, 6.8, H-1'), 5.36 (m), 6.63 (d, 6.0, H-6), 6.66 (s, H-2), 6.75 (d, 7.2, H-4) and 7.13 (ddd, 7.6, 7.6, 2.5, H-5). 13C NMR (CDCl
3,
100.61 MHz): δ14.1 (Me), 22.7, 27.2, 29.3, 29.5, 29.6, 29.7, 30.4, 31.3, 32.0, 35.8 (C-1'), 112.5 (C-6), 115.3 (C-2), 121.0 (C-4), 129.4 (C-5), 129.9 and 130.9, 145.0 (C-3), 155.4 (C-1). For MS data see Table 1.
MixtureIII (1-5, 9, 11, 12, 27, 28, 30, 31)
IR (NaCl) νmax/cm-1: 2919, 2850, 2360, 1587, 1455,
1259, 1151, 1066. 1H NMR (CDCl
3, 400 MHz): δ 0.93 (t,
7.1, Me), 1.31 (bs), 1.62 (m, H-2'), 2.58 (t, 6.9, H-1'), 5.41 (m), 6.69 (dd, 7.6, 2.5, H-6), 6.71 (s, H-2), 6.78 (d, 7.6, H-4) and 7.16 (ddd, 7.6, 7.6, 2.5, H-5). 13C NMR
(CDCl3, 100.61 MHz): δ 14.1 (Me), 22.7, 27.2, 29.4, 29.5, 29.7, 31.3, 31.9, 35.9 (C-1'), 112.6 (C-6), 115.4 (C-2), 120.9 (C-4), 129.3 (C-5), 129.9, 144.9 (C-3), 155.4 (C-1). For MS data see Table 1.
Mixture IV (32, 35, 36, 39-41)
IR (NaCl) νmax/cm-1: 3064, 2917, 2852, 1665, 1607,
1578, 1449, 1315, 1250, 1205. 1H NMR (CDCl 3, 400
MHz): δ 0.89 (t, 6.4, Me), 1.26 (bs), 1.53 (m, H-2'), 2.58 (t, 7.7, H-1'), 3.96 (s, OMe), 5.37 (m), 6.72 (d, 7.6,
H-4), 6.85 (d, 8.0, H-6) and 7.27 (t, 8.0, H-5). 13C NMR
(CDCl3, 100.61 MHz): δ 14.1 (Me), 22.7, 27.2, 29.4, 29.5, 29.7, 29.9, 32.0, 32.1, 36.4 (C-1'), 52.1 (OMe), 111.9 (C-2), 115.6 (C-6), 122.4 (C-4), 129.9, 134.2 (C-5), 146.2 (C-3), 162.6 (C-1), 172.0 (CO). For MS data see Table 1.
MixtureV (29, 32-38)
IR (NaCl) νmax/cm-1: 3064, 2917, 2852, 1665, 1607,
1578, 1449, 1315, 1250, 1205. 1H NMR (CDCl
3, 400 MHz):
δ 0.89 (t, 6.4, Me), 1.27 (bs), 1.52 (m, H-2'), 2.88 (t, 7.8, 1'), 3.95 (s), 5.36 (m), 6.72 (d, 7.2, 4), 6.84 (d, 8.4, H-6) and 7.28 (t, 8.0, H-5). 13C NMR (CDCl
3, 100.61 MHz):
δ 14.1 (Me), 22.7, 27.2, 29.4, 29.5, 29.7, 29.9, 32.0, 32.1, 36.6 (C-1'), 52.0 (OMe), 111.8 (C-2), 115.6 (C-6), 122.4 (C-4), 129.82, 129.84, 134.1 (C-5), 146.2 (C-3), 162.6 (C-1), 172.0 (CO). For MS data see Table 1.
Methylthiolation of mixturesI-V
A 200-500 ng portion (0.4-1.0 nmol) of mixtures I-V was solubilized in 50 µL of DMDS, and 50 µL of carbon disulfide, and 300 µg (1.18 µmol) of iodine were added.
The reaction mixture was kept at 60 °C for 40 h in small-volume sealed vials. The reaction was quenched with aqueous Na2S2O3 (3 × 10-4 mol L-1). The organic phase
was extracted and evaporated to dryness under a nitrogen stream. The residue was dissolved in hexane and analyzed by GCMS.
Acknowledgments
This study was supported by a post-doc grant (SFRH/ BPD/14656/2003) from Fundação para a Ciência e a Tecnologia, Portugal.
References
1. Burkill, H. M.; The Useful Plants of West Tropical Africa, 2th ed., Royal Botanic Gardens: Kew, 1985, vol. 1.
2. Rea, A. I.; Schmidt, J. M.; Setzer, W. N.; Sibanda, S.; Taylor, C.; Gwebu, E. T.; Fitoterapia2003, 74, 732.
3. Gelfand, M.; Mavi, S.; Drummond, R. B.; Ndemera, B.; The Traditional Medical Practitioner in Zimbabwe, Mambo Press:
Zimbabwe, 1985.
4. Ndamba, J.; Nyazema, N.; Makaza, N.; Anderson, C.; Kaondera, K. C.; J. Ethnopharmacol.1994, 42, 125.
5. Mølgaard, P.; Nielsen, S. B.; Rasmussen, D. E.; Drummond, R. B.; Makaza, N.; Andreassen, J; J. Ethnopharmacol. 2001,
74, 257.
6. He, W.; Puyvelde, L. V.; Bosselaers, J.; De Kimpe, N.; Flaas, M. V. D.; Roymans, A., Mathenge, S. G.; Mudida, F. P.; Mutiso, P. B. C.; Pharm. Biol. 2002, 40, 74.
7. Wall, M. E.; Wani, M. C.; Brown, D. M.; Fullas, F.; Olwald, J. B.; Josephson, F. F.; Thornton, N. M.; Pezzuto, J. M.; Beecher, C. W. W.; Farnsworth, N. R.; Cordell, G. A.; Kinghorn, A. D.;
Phytomedicine 1996, 3, 281.
8. Abreu, P. M.; Martins, E. S.; Kayser, O.; Bindseil, K. -U.; Siems, K.; Seemann, A.; Frevert, J.; Phytomedicine1999,6, 187.
9. Ayedoun, M. A.; Moudachirou, M.; Garneau, F. X.; Gagnon, H.; Jean, F. I.; Tomi, F.; Casanova, J.; J. Essent. Oil Res. 1998,
10, 529.
10. Meyer, B. N.; Ferrigni, N. R.; Putnam, J. E.; Jacobsen, L. B.; Nichols, D. E.; McLaughlin, J. L.; Planta Med.1982, 45, 31.
11. Rahman, A.; Choudhary, M. I.; Bioassay Techniques for Drug Development, Harwood Academic Publishers: The Netherlands,
2001.
12. Correia, S. D. J.; David. J. M.; David, J. P.; Chai. H. B.; Pezzuto, J. M.; Cordell, G. A.; Phytochemistry2001, 56, 781.
13. Gonzalez, M. J. T. G.; De Oliveira, C. J. C.; Fernandes, J. O.; Kijjoa, A.; Herz, W.; Phytochemistry1996, 43, 1333.
14. Mata, R.; Calzada, F.; Navarrete, A.; Rio, F. D.; Delgado, G.; J. Ethnopharmacol. 1991, 34, 147.
15. Asakawa, Y.; Masuya, T.; Tori, M.; Campbell, E. O.;
Phytochemistry1987, 26, 735.
16. Yamagima, Y.; Ohashi, K.; Sakamoto, Y.; Hirakawa, S.; Kamikawa, T.; Tetrahedron1987, 43, 3387.
17. Itokawa, H.; Totsuka, N.; Nakahara, K.; Takeya, K.; Lepoittevin, J. P.; Asakawa, Y.; Chem. Pharm. Bull. 1987, 35, 3016.
18. Francis, G. W.; Veland,K.; J. Chromatogr. A1987, 218, 379.
19. Vincenti, M.; Guglielmetti, G.; Cassani, G.; Tonini, C.; Anal. Chem. 1987, 59, 694.
20. Kozubek, A.; Tyman, J. H. P.; Chem. Rev. 1999, 99, 1.
21. Seijas, J. U.; Vázquez-Tato, M. P.; Martínez, M. M.; Santiso, V.; Tetrahedron Lett. 2004, 45, 1937.
22. Jones, T. H.; Brunner, S. R.; Edwards, A. A.; Davidson, D. W.; Snelling, R. R.; J. Chem. Ecol. 2005, 31, 407.
23. Masuoka, N.; Kubo, I.; Biochim. Biophys. Acta 2004. 1688, 245.
24. Muroi, H.; Nihei, K. I.; Tsujimoto, K.; Kubo, I.; Bioorg. Med. Chem.2004, 12, 583.
25. Bouttier, S.; Fourniat, J.; Garofalo, C.; Gleye, C.; Laurens, A.; Hocquemiller, R.; Pharm. Biol.2002, 40, 231.
26. Feresin, G. E.; Tapia, A.; Sortino, M.; Zacchino, S.; Árias, A. R. D.; Inchausti. A.; Yaluff, G.; Rodriguez, J.; Theoduloz, C.; Schmeda-Hirschmann, G.; J. Ethnopharmacol. 2003, 88, 241.
27. Logrado, L. P. L.; Silveira, D.; Romeiro, L. A. S.; Moraes, M. O.; Cavalcanti, B. C.; Costa-Lotufo, L. V.; Ó Pessoa, C.; Santos, M. L.; J. Braz. Chem. Soc.2005, 16, 1217.
28. Kanojia, R. M.; Murray, W.; Bernstein, J.; Fernandez, J.; Foleno, B. D.; Krause, H.; Lawrence, L.; Webb, G.; Barrett, J. F.; Bioorg. Med. Chem. Lett.1999, 9, 2947.
29. Van Beek, T. A.; J. Chromatogr. A2002, 967, 21.
30. Lee, J. H.; Lee, S. Y.; Lee, K. S.; Jang, H. J.; Lee, K. H.; Hahn, T. R.; Park, Y. S.; Planta Med.2004, 70, 1228.
31. Irie, J.; Murata, M.; Homma, S.; Biosci. Biotechnol. Biochem.
1996, 60, 240.
32. Matsumoto, T.; Sei, T.; Agric. Biol. Chem.1987, 51, 249.
33. Hecker, H.; Johannisson, R.; Koch, E.; Siegers, C. P.; Toxicology
2002, 177, 167.