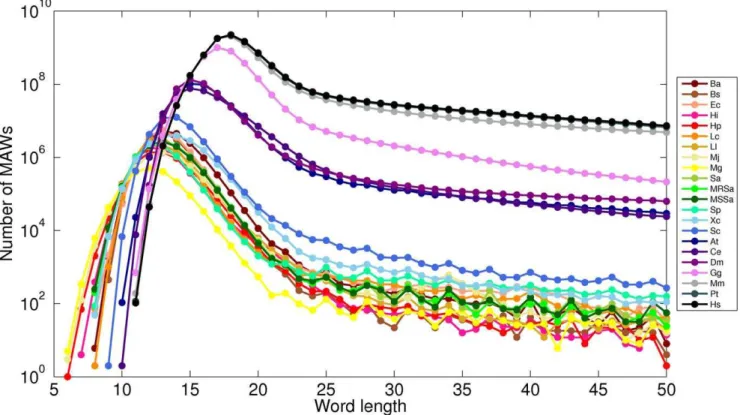
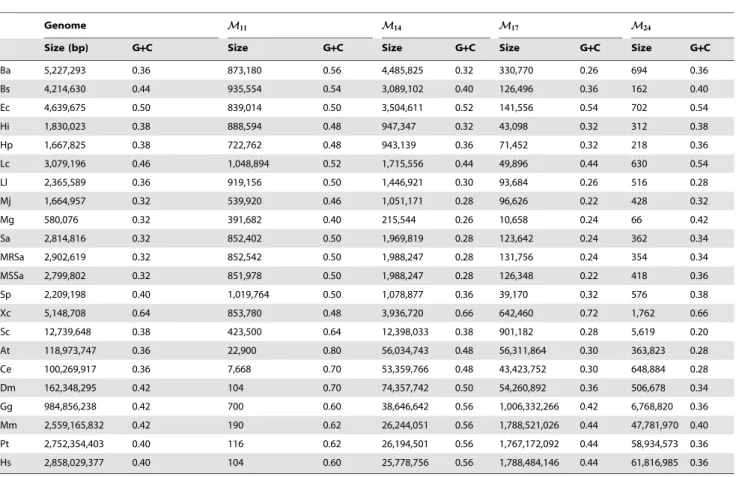
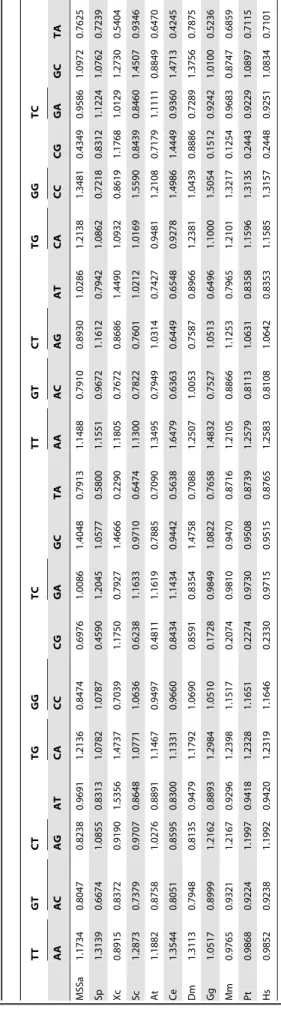
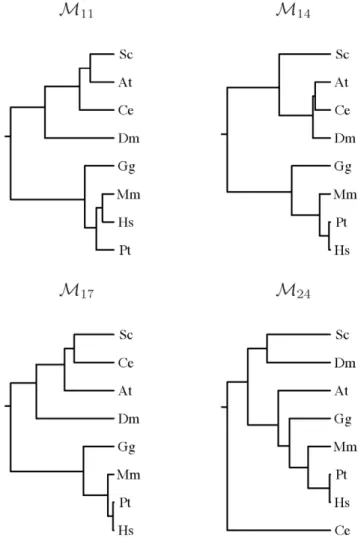
Minimal absent words in prokaryotic and eukaryotic genomes.
Texto
Imagem
Documentos relacionados
Para além do branqueamento da pasta no interior do pulper, também foram recolhidas amostras para determinação da brancura à entrada da torre de branqueamento 1 (ET1), para medir
Extinction with social support is blocked by the protein synthesis inhibitors anisomycin and rapamycin and by the inhibitor of gene expression 5,6-dichloro-1- β-
Uma das explicações para a não utilização dos recursos do Fundo foi devido ao processo de reconstrução dos países europeus, e devido ao grande fluxo de capitais no
O primeiro aspecto é que as diferenças sexuais não podem ser vistas apenas como biologicamente determinadas na acepção meramente genital, antes, são social e
Neste trabalho o objetivo central foi a ampliação e adequação do procedimento e programa computacional baseado no programa comercial MSC.PATRAN, para a geração automática de modelos
Estrutura para análises fisico-químicas laboratoriais 23 Pesca com 'galão' realizada no açude Paus Branco, Madalena/CE 28 Macrófitas aquáticas flutuantes no nude Paus
No caso e x p líc ito da Biblioteca Central da UFPb, ela já vem seguindo diretrizes para a seleção de periódicos desde 1978,, diretrizes essas que valem para os 7
Numa análise individual, observa-se que os valores de tempos de reverberação são bastante diferentes entre museus, sendo que a média das salas do museu londrino é de 2,3 s, o museu
