*Correspondence: R. D. Tanner. Chemical and Biomolecular Engineer-ing Department, Vanderbilt University, Nashville, TN 37235, USA. e-mail: [email protected].
A
rti
Pharmaceutical Sciences vol. 45, n. 4, oct./dec., 2009
Estimating the turnover number in enzyme kinetic reactions
using transient and stationary state data
Sibel Uludag-Demirer
1, Jorge Duran
2, Robert D. Tanner
3*1Villanova University, Civil and Environmental Engineering, Villanova, USA, 2INCYTH/CTUA, Buenos Aires, Argentina, 3Vanderbilt University, Department of Chemical and Biomolecular Engineering, Nashville, USA
Substrate and product concentration data obtained by simulating enzyme-substrate reaction rate equations were used to test two proposed kinetic rate constant estimation techniques in this study. In the irst technique, the turnover number, k3, was calculated using early transient time domain data, which
are dificult to obtain experimentally. The technique used an iterative approach to calculate k3 with a
pair of data and the value of k3 could be retrieved with 35% error. The second technique calculated
k3 using stationary domain data and the value of k3 could be retrieved with less than 5% error. This
second technique also offered internal consistency in the calculation of k3 by calculating k3 both from
the intercept and the slope of the linear plot derived in this study. A series of sensitivity analyses was conducted to understand the robustness of the second technique in estimating k3 from simulated data to
the changes in the reaction rate constants (k1, k2, and k3) and the initial concentration of enzyme used
for simulation. It was found that the second technique generally worked well in the estimation of k3
except for the simulated data for fast substrate conversions such as in the large k3 and [E]0 cases . This
latter method, thus, shows promise for the use of late time experimental substrate/product concentration data to obtain k3. Exclusively using late time data avoids the need for dificult and expensive rapid early
time measurement techniques for estimating k3. Once a reasonable estimate for k3 is obtained, the initial
enzyme value can easily be determined from the maximum velocity constant established from itting the Michaelis-Menten or Briggs-Haldane equations to substrate and product stationary state domain (late time) data. While the irst technique can estimate k3 with only one point in the transient domain,
it is suggested that the second method generally be favored since it only requires late-time stationary domain data and appears to be more accurate.
Uniterms: Enzyme. Kinetics/rate constant. Enzyme reactions.
Dados de concentração de substrato e de produto obtidos por simulação de equações de velocidade de reação enzima-substrato foram usados neste estudo para testar duas técnicas para estimar constantes cinéticas. Na primeira técnica, a constante, k3, foi calculada utilizando os dados de domínio do tempo
inicial de transição, que são difíceis de serem obtidos experimentalmente. A técnica usou uma aproximação iterativa para calcular k3 cujo valor pôde ser estimado com erro de 33%. A segunda técnica calculou k3
usando dados de domínio no estado estacionário e o valor de k3 pôde ser estimado com erro de 5%. Esta
segunda técnica também ofereceu uma consistência interna no cálculo de k3, por calculá-lotanto pela
intersecção quanto pela inclinação da reta derivada deste estudo. Uma série de análises de sensibilidade foi realizada para avaliar a robustez da segunda técnica na estimativa de k3 utilizando dados que foram
simulados quanto às mudanças nas constantes de taxa de reação (k1, k2 e k3) e na concentração inicial de
enzima. Foi encontrado que a segunda técnica, em geral, proporcionou boa estimativa de k3, exceto para
os dados simulados para as conversões rápidas de substrato, como no caso de valores elevados de k3 e de
[E]o. Este último método, portanto, mostra ser promissor quando se usam dados experimentais tardios da
concentração de substrato/produto para obter k3. O uso dados de tempo tardio evita a necessidade do uso
de técnicas difíceis e caras na medida de tempo iniciais para estimativa de k3. Uma vez que é obtida uma
estimativa razoável de k3, o valor inicial da enzima pode ser facilmente determinado a partir da constante
técnica pode estimar k3 com somente um ponto no regime transiente, sugere-se que o segundo método
seja melhor uma vez que ele requer apenas dados do estado estacionário tardio e parece ser mais preciso.
Unitermos: Enzimas. Cinética/constantes. Reações enzimáticas.
INTRODUCTION
The reactions between an enzyme and a substrate are usually analyzed by using the substrate and product concentrations measured over time under isothermal con-ditions. The Michaelis-Menten or Briggs-Haldane model equations are applied to describe such time varying data for the simplest enzyme substrate interactions with the limitation that they exclude the initial (transient) time proiles. The simplest and most often used enzyme subs-trate model is the reaction scheme with one subssubs-trate, one intermediate, and one product with the recovery of the en-zyme (Equation 1) and this model reaction is suficient to describe the behavior of many enzyme-substrate systems (Bernhard, S.,1968).
(1)
In Equation (1), k1, k2, and k3 are the reaction rate constants. In general, the initial substrate concentration ([S]0) is known while the initial (and total) enzyme concen-tration ([E]0) in the reaction system is unknown. Therefore, [E]0 behaves as another parameter to be estimated from the model. Based on the reaction scheme given in Equation (1), the rate expressions of each species follow from the law of mass action:
d[S]/dt=-k1[E][S]+k2[ES] ; [S]=[S]0 (2)
d[E]/dt=-k1[E][S]+(k2+k3)[ES];[E]=[E]0 (3)
d[ES]/dt=k1[E][S]-(k2+k3)[ES] ;[ES]=0 (4)
d[P]/dt=k3[ES];[P]=0 (5)
where [S], [E], [ES], and [P] indicate the molar concentra-tions of the substrate, enzyme, enzyme-substrate complex and product, respectively, in the solution at any reaction time t. Initial concentrations of [ES] and [P] are zero. Typical simulations of [S] and [P] are shown in Figure 1. There are also two independent linear molar conservation equations, as follows:
[E]+[ES]=[E]0 (6)
[S]+[ES]+[P]=[S]0 (7)
Equation (6), an algebraic molar balance on the
en-zyme concentration in it’s two forms, can be obtained by summing Equations (3) and (4) and then integrating, while Equation (7), an algebraic molar balance on the substrate and its products, follows from summing Equations (2), (4), and (5) and then integrating.
The Briggs-Haldane model equation, Equation 8, can be used to it late time (stationary-state) experimental data ([S] and [P]) to calculate the model parameters: the Micha-elis constant (Km) and the maximum velocity term (Vmax).
(d[P]/dt)= v = Vmax [S]/( Km + [S] ) (8)
where Km =(k2 +k3)/k1; Vmax = k3[E]0 and n=product
for-mation rate or velocity
Equation (8) is developed from the stationary-state assumption of d[ES]/dt = 0, and Equation (6). The other widely used correlation equation is the Michaelis-Menten Equation, where the Km term is replaced by Ke = k2 / k1 . That equation is derived by using the equilibrium between E+S and ES, neglecting the subsequent reaction to P and E, along with Equation (6). It turns out that the Briggs-Haldane equation provides a lower bound for d[P]/dt as a function of [S] and the Michaelis–Menten equation an upper bound as a function of [S] (Rutherford, B.J. et al., 2007).To calculate the model parameters, a nonlinear regression analysis can be performed. Typically, a linear arrangement of reciprocal n (product formation rate)
plot-ted against reciprocal [S] in the form of a Lineweaver-Burk plot is generally preferred for recovering the model para-meters. To calculate the initial concentration of enzyme, [E]0, k3 should be estimated separately.
FIGURA 1 - Simulated concentrations of substrate and product
for a reaction medium containing [S]0 = 1, [E]0 = 0.1 relative
The turnover number, k3, can be determined directly from Equation (5), if both [ES] and [P] are measured at specified time intervals. Usually, however, [ES] is not measured, and k3 must then be determined from the early time (transient) data from measurements of [S] and [P] at speciic time intervals. [ES] is generally not very accurate if obtained from [S] and [P] measurements and [S]0 using Equation (7), since it is the small difference between large numbers with relatively large measurement error. Assu-ming that at very early reaction times, [P] is negligible with respect to [S]0, i.e., [P] → 0, then Equation (7) reduces to:
[ES] = [S]0 - [S] (9)
Now, replacing [ES] in Equation (5) with (9), Equa-tion (10) can be obtained as previously shown (Tanner et al., 1977).
d[P]/dt = k3 ([S]0 - [S]) (10)
With the measurements of product and substrate concentrations corresponding to the transient period, it is possible to determine k3 for [S] ≤ [S]0 from the slope
of the expression in Equation (10) by plotting d[P]/dt against ([S]0 – [S]) for a positive slope or against [S] for a negative slope, as pictured by the line (qualitatively) near the peak in Figure 2. The estimated value of k3 from Equation (10) increases as [S]→[S]0, so that the tangent
at [S]0 gives the most accurate value for k3. However, it is dificult to obtain the early time product formation rates since enzyme-substrate reactions are generally too fast, and there are typically only one or two data points taken (at best) in the transient period. Measurement errors come into play as well. Since [S]0 – [S] is small in early time, the variance associated with [S] dominates this differen-ce and becomes controlling. It is noted that, without the assumption of small [P], Equation (10) would be replaced by: d[P]/dt = k3([S]0 - [S] –[P)). This rigorous replacement equation will be invoked in the iterative method which is described in next section.
Turnover Number Estimation Using The Maximum Product Formation Rate
Numerically integrating the model given by Equa-tions (2) through (5), employing ki = 1 (i = 1, 2, 3), [E]0 = 0.1, [S]0=1 in dimensionless molar units and using a linear, triangular, implicit integration scheme for a step size of 0.01 time units up to 90 time units leads to the set of lated data presented in Figure 1 (Tanner, 1972). The simu-lated data, accurate to three decimal points, can be divided
into two domains: transientand stationary state (Figure 2). The transient domain extends from the initiation of the reaction (t = 0; [S]=[S]0), to the moment (t=tpeak) when the maximum value is registered for the product generation rate (d[P]/dtpeak at [S] = Speak ). The stationary state domain includes the data from t=tpeak to t=∞, i.e., from d[P]/dt peak
to the end of the reaction, d[P]/dt=0 as t→∞ and [S] →0
(Figure 2). Typically,there is only a single data point simulated in the transient state domain when [E]0 equals 1/10 of [S]0 if the point at the boundary between transient and stationary state is excluded (Tanner, 1972).
In this section, the estimation of k3 with only one pair of data points is illustrated using an iterative technique. Since special equipment like the stopped low apparatus is generally required for multiple measurements of [S] and [P] in the transient state domain, it is not always possible to use Equation (10) to estimate k3. In addition, it is dificult to precisely deine the maximum rate of product formation d[P]/dtpeak with a corresponding S value of Speak) to deine the transient state domain precisely and then to accurately use it with the initial time point, [S]=[S]0 and d[P]/dt=0 to estimate k3. Therefore, an iterative procedure should be de-veloped to determine d[P]/dtpeak and k3. A point near d[P]/ dtpeak and Speak is assumed to be the largest estimated d[P]/dt value and is used as the starting point for the iteration. The pair of d[P]/dtpeak(pointed out in Figure 2) and Speak measu-rements can then be used to draw the line connecting the peak and the origin (below the slope as the transient line shown in Figure 2) and k3 can be calculated from the slope of the line. The irst iteration in this example provided a k3 estimate of 0.45 in reciprocal time units compared to 1, the value originally employed in the simulated model. In an effort to improve the accuracy of this calculation, an iteration procedure is developed to sharpen the estimate of the value of the slope. Since the most accurate slope in
FIGURA 2 - A cross plot of the data in Figure 1 for the
estimation of the turnover number (K3) using the maximum
the transient domain is the tangent at [S]=S0 and since this is equal to k3, the negative slope has to be steeper than that of the line connecting the peak and the initial value, such as the slope of the line in the transient domain pictured in Figure 2. This slope has a value of -(k3)2 for the second iteration, in which the subscript stands for the number of iterations, as shown in Figure 2. The subscript or k3 refers to the second iteration. To enhance the convergence of the results, an “overshoot segment” is deined as the product of k3 and Ppeak and it was added to the d[P]/dt | peak
previou-sly employed to determine k3. It is hypothesized that this additive term will bring the value of d[P]/dt | peak closer to
the desired d[P]/dt value on the initial tangent line. The theoretical development of the method and the iteration sequence now follows (in the six indicated parts):
• Initial estimate of k3
Initial estimate of k3 starts with using Equation (10). This equation was derived by assuming that [P] in Equation (7) is small with respect to [S]0 - [S]. (d[P]/dt)0 represents a real experimental value, which is typically at the largest [S] value in the stationary state domain, and it is equal to the observed (d[P]/dt)peak.. It is the starting rate in the beginning iteration. The value of [S]peak, which is the substrate concentration corresponding to the maximum product formation rate, will remain the same at each step of the iteration. It therefore follows that the irst estimate for k3 is:
(k3)1 = (d[P]/dt)0 / ([S]0 - [S]peak), (11)
where the subscript 1 indicates the irst iteration.
Note that the slope is negative for the tangent line in Figure 2 since the d[P]/dt value is zero at the point of initial substrate. The two [S] terms in the denominator of Equation (11) are switched from the usual order for a tangent line in order to make (k3) positive in Equation (11).
• Calculation of the overshoot segment using Equations of (5) and (7):
Since: (d[P]/dt)1 = (k3)1([S]0 - [S]peak) + (k3)1[P]peak, the overshoot segment for the calculation of (d[P]/dt)1 is equal to (k3)1 [P]peak, where [P]peak is the concentration of product at the maximum product formation rate. [P]peak like [S]peak remains constant throughout the iteration.
• Deinition of the overshoot point or (d[P]/dt)2 :
The irst iterative equation thus becomes:
(d[P]/dt)1 = (d[P]/dt)peak + (k3)1[P]peak (12)
• Calculation of the next estimate of k3:
(k3)2 = (d[P]/dt)1 / ([S]0 - [S]peak)
• Calculation of the new overshoot segment:
(overshoot segment)2 = (k3)2 [P]peak
• Calculation of the new overshoot point:
(d[P]/dt)2 =(d[P]/dt)peak + (k3)2 [P]peak
In general, we have two iteration equations as follows:
(k3)n = (d[P]/dt)n-1/([S]0-[S]peak) (13) (d[P]/dt)n = (d[P]/dt)peak + (k3)n [P]peak (14)
where n is the iteration number. Here, (d[P]/dt)peak was a ixed number. This iterative procedure is performed until the value of k3 appeared to converge. The iteration proce-dure was stopped when the retrieved value of k3 was within a difference of ± 0.01.
This procedure was applied to the simulated data (Figure 2) and the value of k3 appeared to converge to 0.65 after five iterations, but clearly more iterations would have been in order since the known outcome of 1 should be reached.. This procedure retrieved the value of k3 with a 35% error. Combining Equations (13) and (14) and generalizing for the n’th case gives: (d[P]/dt)n = (d[P]/dt)peak [1+x+(x)(x)+…] = /(d[P]/dt)peak (1-x) for x less than 1, in this geometric progression. Here, (x= Ppeak/ ([S]0-[S]peak), which is less than 1, since [P]/([S]0-[S] ) is less than 1 in general from (Equation (7) because [ES] is positive). Thus, (d[P]/dt)n converges so that Equation (14) converges for large n . It is noted that the iterative value of k3 in this procedure used only a single data point. Stopping at five iterations was in keeping with another iterative procedure in the transient domain in which three to ive iterations typically resulted in the desired outcome. That procedure was based on the use of orthogonal polynomials and Picard’s iteration technique and used essentially the same simulated data (Tanner, R.D.,1972). Since there is only one data point in this method, another technique was explored for the estimation of k3 using the much more accessible stationary state domain data (See Figure 2).
Turnover Number Estimation Using Stationary State Data
domain, and without requiring the dificult-to-obtain tran-sient data, although such additional data would enhance the itting technique. Starting with Equation (5), the [ES] complex concentration can be expressed in terms of [P] and [S] employing Equation (7) to obtain:
(16)
After division of Equation (16) by [P] the following mathematical expression is obtained:
(17)
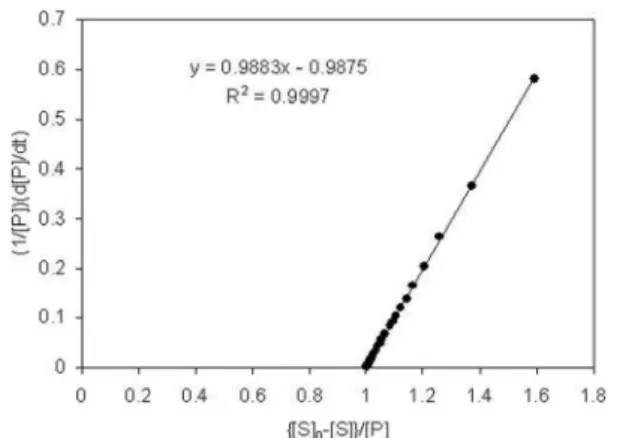
This form of the product formation rate equation is very helpful in estimating the k3 constant, because it bears a linear relationship between 1/[P] d{P}/dt and ([S]0 – [S])/ [P] and k3 can be calculated both from the intercept and the slope of the plot. Linearity in Equation (17) may also be useful in directly testing the original premise shown in Equation (1), as stated in Equations (2)-(5). An example of such a linear plot is shown in Figure 3 for [S] and [P] simulated data from Figure (1) with 3 signiicant igures. Note that as t →∞, the line goes to the point (1,0 ). This is seen, in general, for the “x” coordinate going to one as [S] goes to zero and [P] goes to [S]0 as t →∞ and for the “y” coordinate going to zero as [P] goes to [S]0 and d[P]/dt goes to zero as t →∞. Internal consistency of these two estimated k3 values serves as a check on the scatter and the time spacing of the [S] and [P] data as well as the integrity of the differenced value, D[P]/Dt, which is used to
estimate d[P]/dt. The linear regression analysis of the plot shown in Figure 3 resulted in four place k3 values as 0.9883 using the slope and 0.9875 using the intercept. The goodness of it of the method could be evaluated in two ways: (a) Accuracy, the difference with the known parameter value used to simulate the data, and (b) Consistency, the internal difference between the slope and intercept estimates of k3. The irst one is related to the accuracy of the method that is the average of the k3 values, 0.988 (in keeping with the three place simulated data), was found to be very close to 1, the value used to create [S] and [P]. The difference with the known value for this particular regression was measured considering the percent difference between the average k3 calculated and the known k3 value and estimated as 1.24%, which is close to the desired 0% for highest accuracy. The internal difference was evaluated by calculating the percent difference between the slope and the intercept values for k3 and estimated as 0.08%, which is negligibly small as compared to a desired 0% for desired internal consistency.
The two parameters in the Briggs-Haldane model equation, Equation (8), Vmax and Km, are generally estima-ted using stationary state data with the Lineweaver-Burk expression, Equation 18). This equation is the reciprocal of Equation (8) and is used to formulate a linear form to use as an alternative to the nonlinear regression of the hyperbolic Equation (8):
(18)
In estimating the Km and Vmax constants with the Lineweaver-Burk plot, linear regression analysis of the straight line was performed (Figure 4). The values of the parameters are calculated as 0.968 concentration units and 0.0588 moles per unit time for Km and Vmax respectively. Since Vmax = k3 [E]0, the initial concentration of the enzy-me, which is typically unknown in the system, was calcu-lated as 0.06 concentration units which is different by 40% from the product of the original parameter values used to
FIGURA 3 - The estimation of the turnover number, K3, from
the slope and intercept using three place stationary state domain data obtained from the simulation underlying Figures 1 and 2. The smallest time point is closest to the upper right corner.
FIGURA 4 - Lineweaver-Burk Plot of the simulated data. The
simulate in the differential equations. The Km estimate was off by 50%. These are large errors when compared to those incurred in estimating k3 from Equation (17).
Sensitivity Analysis
The enzyme-substrate single intermediate model previously introduced in Equation (1) contains four typi-cally unknown parameters; k1, k2, k3, and [E]0, given [S]0. Here, the accuracy of the parameter estimation method comparing Equation (17) to simulated data in determi-ning k3 and its sensitivity to the variation of the different parameters used in simulating the data is evaluated. Also considered in this analysis is the effect of the number of signiicant igures employed in the linear regression procedure.
The computer software, Polymath, was employed to simulate Equations (2) through (5) in order to determi-ne the evolution of substrate and product concentration trajectories with time by using an implicit modiied Euler method. These simulated data were then used to estimate the value of k3 both from the slope and intercept using Equation (17). The estimated value of k3 for a particular it was then calculated by averaging the k3 values obtained from the slope and from the intercept. Only the data cor-responding to the stationary state domain, following the (d[P]/dt)peak for late times, were utilized in the proposed itting Equation (17). This limitation to only stationary state data was followed even though Equation (17) applies in general to include the early time transient as well as the stationary data, because in practice only stationary data are readily available.
The goodness of it of the method was evaluated in terms of accuracy and consistency. The absolute difference with the known value, k3, for a particular regression was determined to develop the accuracy using the percent di-fference between the average k3 calculated and the known k3. The internal difference was evaluated by calculating the percent difference between the slope and the intercept values for k3, to determine the consistency of the method.
Effect of k1 variation on the estimate for k3
Values of 0.01, 0.1, 1, and 10 for k1, which were used to simulate three place data for the estimation process, were selected in this part of the study to estimate the effect of k1 variation on k3 estimation. The remaining parameters, k2, k3, [E]0, and [S]0 were kept equal to 1, while the initial product concentration [P]0 was kept at zero throughout these case studies. Note that [E]0 is now 1 rather than 0.1 as in Figure 1.
When k1 was larger than 1, the percent absolute di-fference of the k3 value determined with the linear regres-sion (Equation 17) with respect to the known value was generally unacceptable (above 10% error). However, for k1 equal to 0.1 or less, that difference decreased to levels less than 10% (Figure 5). Equation (7) itself is useful in helping us understand why the absolute value of the estimated k3 increases as the value of k1 increases. Equation (7), when divided through by [P] yields Equation (19):
(19)
It can be seen, at least for late time data using Equation (8), that as Km =(k2 +k3)/k1 increases goes towards zero and (dP/dt) increases. This means that the rate at which (P) is formed increases, up to (P) =1 at t=∞. This is when [P] goes
to [S]0=1, when (ES) and (S) go to zero referring to Equation (7). As t approaches ∞, the term (dP/dt) in Equation (17)
eventually tends towards zero since d[P]/dt = k3 [ES] and [ES] goes towards zero while [P] goes to [S]0 . Thus, the term ([S]0 – [S])/[P] drives to 1. This is also observed in Figure 3. and can serve as in internal check step for itting Equation (17) as seen in Figure (3).
Since higher k1 values correspond to higher rates of substrate consumption, the number of spread out data points available for a reasonable least squares it of the data decreases (in both the substrate and product domains) affecting the accuracy of the linear regression. For very large k1 , for example, the points tend to collapse to one point (zero for [S] and one for [P] ). With fewer data points in hand, it can be seen that it would be more and more dificult to obtain both accurate slope and intercept values
FIGURA 5 - Percent absolute difference of the k3 value
from Figure 3, since the early time high [S] and low [P] values tend to deine the entire itting domain. Interes-tingly, the internal consistency between the k3 estimated from the slope and the k3 from the intercept becomes more acceptable as k1 approaches 10 (Figure 6).
Effect of k2 variation on the estimate for k3
Four simulations were performed with k2 values at 0.01, 0.1, 1 and 10 while k1, k3, [E]0, and [S]0 were kept equal to 1, keeping [P]0 at zero. The percent absolute differences of the estimated k3 values with the known value, 1, were very high for the k2 range tested in the study (Figure 5). The simulation performed with the lowest k2 value (0.01) yielded only two data points to estimate k3 using Equation (17) due to the fast conversion of substrate to product in the early transient domain with this small back reaction term. Moreover, there was very poor internal consistency in the calculation of k3 from the slope and the intercept of Equation (17). Therefore, the percent absolute difference is not shown for this case in Figure 6. Interestingly as the reverse reaction became more important (larger k2), there was a decrease in the absolute difference of k3 from the known value as the value of k2 increased, from 0.1 to 10 (Figure 5). This decrease was due to the increase in the number of data points spread over the entire stationary state domain. At the same time, the internal difference values of k3 obtai-ned from the slope and the intercept of the Equation (17) became insigniicant as shown in Figure 6.
Effect of k3 variation in the equations used to simulate the data on the estimate for k3 from those data
Again, four different values in the range of 0.01-10 for the turnover number, k3, were selected to test the effect of the magnitude of k3 on the performance of the method in predicting the known value. The remaining parameters, k1, k2, [E]0, and [S]0 were kept equal to 1, while [P]0 was made equal to zero. The absolute value of the difference between the k3 determined with Equation (17) and the known value remained below 0.5 % for k3 values equal or lower than 0.1, indicating good accuracy of the method in that range. However, the difference with the known value increased for higher k3 values, reaching, for instance, 80 % for k3 equal to 1 (Figure 5), showing that the method becomes signiican-tly less accurate at high k3 values. An explanation for this breakdown in the estimations is that there is the lack of data in the stationary state domain for these cases with high k3 values and this leads to large itting errors. This lack of data points is the direct result of having the conversion rate of the substrate become so fast that the product concentration reaches 1 concentration unit rapidly in the early transient domain with the reaction essentially reaching completion. Thus there are few remaining data points left for itting in the stationary state domain for itting when these early time points are not measurable in practice.
It is observed that the internal difference is negli-gible in the estimations of higher k3 values (Figure 6). The internal difference is calculated as 27% for the case where the k3 value is 0.01. This is the lowest value tested in the study, indicating that at low k3 values (i.e. 0.01) the consistency of the method tends to break down leading to a high variance in the estimated value for k3.
Effect of [E]0 variation on the estimate for k3
The initial concentration of enzyme ([E]0) was varied in four simulation runs, adopting values of 0.01, 0.1, 1, and 10 concentration units. The remaining parameters, k1, k2, k3, and [S]0 were kept equal to 1, while [P]0 was kept equal to zero. When [E]0 had values equal to or smaller than 1, the percent difference of the k3 value estimated with Equation (17) and the known value remained within 10% (Figure 5). At a high [E]0 concentration (10 concentration units), that difference started to increase, reaching 191%. It seems that at very high initial enzyme concentrations the accuracy of the method deteriorates due to the very fast consumption of the substrate available in the system and lack of ittable data in the stationary-state (late time) domain. The resulting fitting is like putting a line through one point. The high
FIGURA 6 - Internal consistency of the turnover estimation
method using Equation (17) and the known given values of k1,
k2, k3, and E0 (shown above) used to simulate the data used for
internal consistency of the method was found to be almost independent of the initial enzyme concentration (Figure 6).
Inluence of the number of signiicant igures employed
Analytical results are often expressed with two sig-niicant igures rather than with the three sigsig-niicant igure data employed in the above sensitivity analyses. In order to observe the variation introduced in the previous [E]0 sensi-tivity analysis employing more “standard quality” data, the values originally employed were rounded to two signiicant igures. As expected, the trends observed in absolute values were similar, but their magnitudes increased for each case. An absolute difference of 12.2% with the known value of k3 was observed with the two signiicant igure substrate and product concentration data compared to the absolute difference of 11.6% with the known value of k3 substrate and product concentration data with ive signiicant igures.
CONCLUSIONS
Two kinetic rate estimation techniques were studied using simulated data of substrate and product concentra-tions. The data were obtained by integrating the standard enzyme-substrate nonlinear differential equations (Equa-tions 2-5) with the rate constants (k1, k2, k3) equal to 1 and [S]0=1 = [E]0 . The turnover number, k3, was calculated in the irst method using early transient domain data with an iterative method. k3 was found to equal 0.65 in this method after ive iterations. This value lies within the bound of 0.4 k32.1estimated in an earlier study using the same data and a simple polynomial itting technique (Tanner, 1972). A second method for the estimation of k3 was developed using stationary-state (late time) data only. The sensitivity of this method to changes in the reaction rate constants, including the turnover number, and the initial enzyme con-centration was tested. It was clear that the method broke down in those cases of high rates of substrate consumption corresponding to very high enzymatic levels, a relatively unusual situation in traditional enzyme kinetic experi-ments. When all of the substrate is converted to product ([P]max = 1, [S]0=1 ) in the transient domain, there are no or limited data in the subsequent in time stationary domain to be used in the linear regression calculations. However, the method yielded very good estimates of k3 in these cases containing suficient data in the stationary phase. By its generally good accuracy for all but the highest k3 and very high enzyme level cases the applicability of this method now provides a good reason for measuring both substrate and product concentration data in experimental
enzy-me kinetic systems in order to estimate k3, the turnover number, directly. Once k3 is established, the total enzyme concentration is readily calculated from the Vmax term of the Briggs-Haldane or the Michaelis-Menten equation.
Estimating k3 from late-time data in this new method, described by Equation (17), obviates the need for dificult-to-obtain initial rate data. The required late-time data or substrate and product concentrations needs to be of such quality as to provide reasonably accurate product rates, d[P]/dt, and normalized product rates, (1/[P])d[P]/dt, as well as the substrate difference, [S]0 - [S] and its division by [P]. This new method for estimating k3 not only provi-des a relatively simple and easy way to estimate [E]0, but when k1 and k2 are estimated by other techniques, it offers the parameters to directly validate the original differential equations, Equations (2) and (5), against experimental data.
ACKNOWLEDGEMENT
We thank one of the reviewers of this paper for the comment:
“Most biochemists employ the method of initial rates to obtain steady-state data for parameter estimation (Briggs-Haldane equation). Complete progress curves are valuable for biochemists to generate, in addition to initial rate data, because comparison of the itted parameters can reveal anomalies for further study.”
REFERENCES
BERNHARD, S. The structure and function of enzymes. New York: W.A. Benjamin, 1968. p.73.
RUTHERFORD, B. J.; HAHS, D.; TANNER, R.D. Quantifying the position of the stationary state trajectory between the Michaelis-Menten and Briggs-Haldane equations as a function of the initial enzyme concentration. Chem. Eng. Comm., v.194, n.7, p.867-888, 2007.
TANNER, R.D. Estimating kinetic rate constants using orthogonal polynomials and Picard’s iteration method. Ind. Eng. Chem. Fundamentals, v.11, n.1, p.1-8, 1972.
TANNER, R. D.; LOO, A. C.; SHISLER, J. L.; REED, M. W.; ROWLETT, R. D.; MORRIS, J. W.; SCHLOSSNAGLE, G. W.; OVERLEY, J. R. Mapping the lag phase and bounding the growth phase in fermentation reactions. AIChE Symp. Ser., v.78, n.167, p.55-65, 1977.
Received for publication on 12th June 2009.
![FIGURA 1 - Simulated concentrations of substrate and product for a reaction medium containing [S] 0 = 1, [E] 0 = 0.1 relative normalized molar units with rate constants of k i = 1 (i = 1,2,3).](https://thumb-eu.123doks.com/thumbv2/123dok_br/15412127.586619/2.892.486.796.240.434/simulated-concentrations-substrate-reaction-containing-relative-normalized-constants.webp)