Printed in Brazil - ©2007 Sociedade Brasileira de Química 0103 - 5053 $6.00+0.00
Article
*e-mail: [email protected]
GC-MS Determination of Organochlorine Pesticides in Medicinal Plants Harvested in Brazil
Marili V.N. Rodrigues,a Felix G. R. Reyes,b Pedro M. Magalhãesa and Susanne Rath*,c
a
CentroPluridisciplinar de Pesquisas Químicas, Biológicas e Agrícolas, Universidade Estadual de Campinas, CP 6181, 13083-970 Campinas-SP, Brazil
b
Faculdade de Engenharia de Alimentos, Universidade Estadual de Campinas, CP 6121, 13083-970 Campinas-SP, Brazil
c
Instituto de Química, Universidade Estadual de Campinas, CP 6154, 13084-971 Campinas-SP, Brazil
Neste trabalho foi desenvolvido um método para a determinação de hexaclorobenzeno, lindano,
heptaclor, heptaclor epóxido, aldrin, dieldrin, endrin, DDT e DDE em folhas de Mikania laevigata,
Maytenus ilicifolia e Cordia verbenacea, empregando a Cromatografia Gasosa acoplada a Espectrometria de Massas (GC-MS). A extração dos pesticidas foi realizada por extração sólido-líquido (SLE), seguida de limpeza por extração em fase sólida (Florisil e silica-gel). A quantificação foi realizada usando o GC-MS no modo SIM. A recuperação média variou de 70 a 124%. A
precisão inter-ensaio de uma amostra fortificada com 200 ng g-1 de cada pesticida variou de 1,0 a
7,3%. Os limites de quantificação variaram de 3,0 a 30 ng g-1 e estão abaixo dos limites máximos
de resíduos (MLR) estabelecidos para os pesticidas sob estudo. O método foi empregado para
analisar amostras de Mikania laevigata, Maytenus ilicifolia e Cordia verbenacea de um campo
experimental de Paulínia, SP, Brasil. As amostras apresentaram contaminação com dieldrin acima
do MRL estabelecido pela Farmacopéia Européia (50 ng g-1).
A method using Gas Chromatography coupled to Mass Spectrometry (GC-MS) for the determination of hexachlorobenzene, lindane, heptachlor, heptachlor epoxide, aldrin, dieldrin,
endrin, 4,4’-DDT and 4,4’-DDE in leaves of Mikania laevigata, Maytenus ilicifolia and Cordia
verbenacea was developed. Extraction of the pesticides was carried out by solid-liquid extraction (SLE), followed by clean-up in solid phase mixed cartridge (Florisil and silica-gel). Quantification was performed using GC-MS in the selected ion monitoring mode. Mean recovery rates of 70
to 124% were obtained. The inter-assay precision of a sample fortified with 200 ng g-1 of each
pesticide was in the range of 1.0 to 7.3%. The quantitation limits ranged from 3.0 to 30 ng g-1
and were below the Maximum Residue Limit (MRL) for all the pesticides under study. The
method was employed to analyze samples of Mikania laevigata, Maytenus ilicifolia and Cordia
verbenacea from an experimental field in Paulínia, SP, Brazil. The samples presented contamination with dieldrin above the MRL established by the European Pharmacopoeia (50 ng g-1).
Keywords: organochlorine pesticides, medicinal plants, GC-MS, Mikania laevigata, Maytenus ilicifolia,Cordia verbenacea
Introduction
Medicinal plants or their derived material, have been widely employed in all cultures, throughout history, for the prevention and treatment of diseases. The significant increase in the use of herbal medicines in recent decades may be attributed to popular wisdom, the costs of synthetic
drugs and the resurgence of interest in the development of new drugs and the re-establishment of old ones from plant sources.
136 GC-MS Determination of Organochlorine Pesticides J. Braz. Chem. Soc.
potential adverse health effects for consumers and seems to be overshadowing the potential genuine health benefits of herbal products.1
Like other vegetables, medicinal plants are composed of many constituents and are of great variability due to different growth, harvest, processing and storage conditions. Furthermore, similar to other agricultural products, there could be contamination, from micro-organisms, heavy metals and pesticides. The conta-mination of medicinal plants by pesticides may be a consequence of the use of pesticides during cultivation, migration from neighboring cultures or due to environ-mental contamination.2
Organochlorine pesticides (OCP) were widely used during the last century for agricultural and non-agricultural purposes throughout the world. Even though they have been banned in most countries (in Brazil since 1985), due to their high persistence in the environment, their residues are still found in many matrices, including medicinal plants.3
Several analytical methods for the determination of pesticides in different matrices, like foodstuffs, water and soils, are reported in the literature, where chromatographic methods are the most widely applied.4-7
Although highly efficient chromatographic methods have been developed, it is not possible to establish a single analytical method for multiresidue analysis because of the large number of pesticides available, the low level of contamination and the diversity of matrix composition.
Analytical methods established for pesticides in foodstuffs, as well as in other matrices, can not be unconditionally applied to phytopharmaceuticals without prior validation. In general, the analytical method involves the following stages: sampling, sample preparation, extraction, detection and data analysis, where the sample preparation could be considered a critical step. The official multiresidue pesticide methods recommend extraction of the pesticides with organic solvents,8 clean-up by column
chromatography4,9,10 or gel permeation chromatography8,11
and quantification by capillary GC-MS.12-14 Since the
recommended extraction procedure requires large quantities of toxic solvents, other extraction procedures have been reported, including, solid-phase extraction,9,10,15,16
supercritical fluid extraction17 and microwave assisted
extraction coupled to solid phase microextraction.18
With relation to current Brazilian legislation, which regulates the registration of phytopharmaceuticals, the determination of pesticides has not been included in the purity assay of herbal plants.19
Whereas the European Pharmacopoeia presents the MRL for several pesticides in medicinal plants, including
organochlorine pesticides, the Brazilian Pharmacopoeia neither defines the MRL, nor recommends analytical assay for these contaminants. Nevertheless, the fact that they are not included in the Pharmacopoeia does not mean that they should be tolerated.3
Even though there have been several papers published related to the presence of OCP in medicinal plants, there are only a few reports on the occurrence of OCP in medicinal plants harvested in Brazil.20 Therefore, the
objective of the present work was the optimization and validation of a simplified method employing SLE and capillary GC-MS for the determination of hexachlorobenzene, lindane, heptachlor, heptachlor epoxide, aldrin, dieldrin, endrin, DDT and DDE (Figure 1) and the application of this methodology for quantifying OCP in Mikania laevigata, Maytenus ilicifolia and Cordia verbenacea, medicinal plants extensively commercialized
in Brazil. The leaves of some species of Mikania
(Asteraceae) are widely used to treat respiratory tract
diseases, whereas the species of Cordia verbenacea
(Boraginaceae), popularly known as black sage, are used for the treatment of several inflammatory processes and as a healing agent. The fresh or dry leaves of Maytenus ilicifolia (Celastraceae) a native plant in Brazil, popularly known as congorosa, are used to alleviate stomach pain and nausea and to treat ulcers and gastritis.
Experimental
Chemicals
All the reagents used were, at least, of analytical grade. Hexachlorobenzene (99%), lindane (98.9%), heptachlor (99.5%), aldrin (98.5%), heptachlor epoxide (99.5%), dieldrin (98.9%), 4,4’-DDE (99.3%), endrin (99.0%) and 4,4’-DDT (98.8%), purchased from Chem Service (West Chester, PA, USA), were used for the preparation of standard solutions. For the solid phase extractions, Florisil (60 - 100 mesh) and silica gel 60 (0.063 – 0.200 mm) from Merck (Rio de Janeiro, Brazil), and residue grade anhydrous sodium sulfate and HPLC grade dichloromethane from Mallinckrodt Baker (Phillipsburg,
NJ, USA) were used. Residue grade n-hexane was
purchased from Tedia (Fairfield, OH, USA).
Equipment
capillary column HP-5 (30 m x 0.25 mm I.D.; 0.25 µm film thickness) (Folsom, CA, USA). The GC-MS interface
temperature was 300 oC. Samples were injected
automatically by means of an HP 7673 autosampler in splitless mode with the split outlet opened after 1.5 min with injector port temperature at 270 oC. The helium
carrier gas flow rate was 1 mL min-1. The column
temperature program was as follows: 90 oC, held for 1
min, 12 oC min-1 to 150 oC, held for 1 min, 2 oC min-1 to
230 oC, held for 3 min, 10 oC min-1 to 275 oC, held for 25
min.
The selected ion monitoring (SIM) mode was used in quantification analysis. The mass-spectrometer acquisition settings were: electron-impact ionization 70 eV, solvent delay 10 min, electron multiplier voltage 2000 V. A SIM table was constructed for GC-MS quantification. Three specific ions (the most abundant as quantification ion and
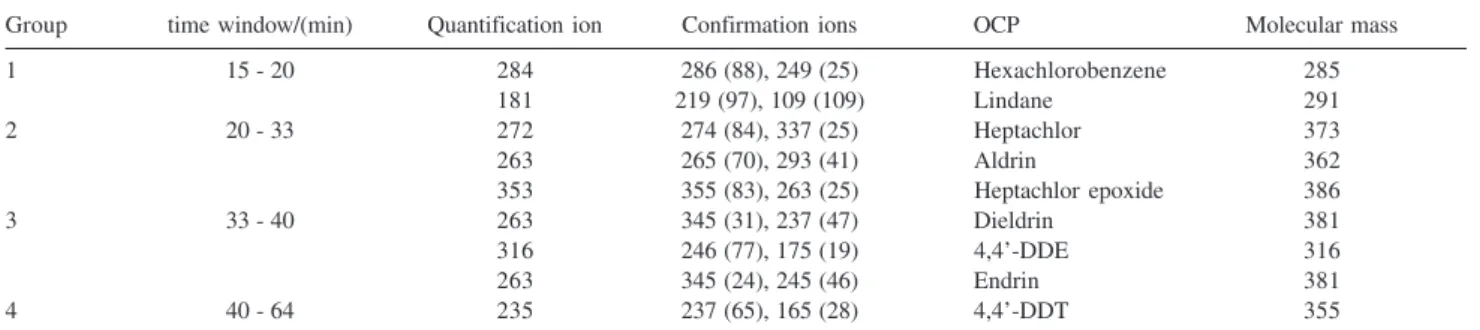
two ions for confirmation) were monitored for each pesticide (Table 1). Retention time and abundance of the confirmation ions relative to that of quantification ion were used as identification criteria.
Samples
Samples of Mikania laevigata Schultz Bip (20 plants, voucher code UEC 102.046), Maytenus ilicifolia Mart.
ex Reiss (22 plants, voucher code UEC 112.745) and
Cordia verbenacea D.C. (18 plants, voucher code UEC 112.774) were collected on 14 January 2002 in the experimental area of the Chemical, Biological and Agricultural Research Center (CPQBA) of the State University of Campinas located in the city of Paulínia, SP, Brazil. The vouchers of the specimens were deposited at the herbarium of the Department of Botany of the
Figure 1. Chemical structures of the OCP.
Table 1. Settings used for data acquisition by selected ion monitoring (SIM)
Group time window/(min) Quantification ion Confirmation ions OCP Molecular mass
1 15 - 20 284 286 (88), 249 (25) Hexachlorobenzene 285
181 219 (97), 109 (109) Lindane 291
2 20 - 33 272 274 (84), 337 (25) Heptachlor 373
263 265 (70), 293 (41) Aldrin 362
353 355 (83), 263 (25) Heptachlor epoxide 386
3 33 - 40 263 345 (31), 237 (47) Dieldrin 381
316 246 (77), 175 (19) 4,4’-DDE 316
263 345 (24), 245 (46) Endrin 381
4 40 - 64 235 237 (65), 165 (28) 4,4’-DDT 355
138 GC-MS Determination of Organochlorine Pesticides J. Braz. Chem. Soc.
Institute of Biology of the State University of Campinas (UNICAMP), SP, Brazil.
In addition, two samples of Mikania sp were purchased at a retail market in Jundiaí, SP, Brazil.
The plants were dried at 40 °C for 72 h in an air recirculation oven, model Blue M (Blue Island, IL, USA), the stalks removed and the leaves grounded into a fine powder in a knife mill, MR Model MMR 320 from Manesco & Ranieri (Piracicaba, São Paulo, Brazil) and stored in a freezer (-18 °C) until analysis.
Analytical procedure
The extraction of the pesticides from the solid samples (2.0 g) was carried out by solvent extraction with 15 mL of
n-hexane:dichloromethane (4:1 v/v). For this purpose, the sample was blended in an Ultra-Turrax system (Zurich, Switzerland) for 3 min and then the material was transferred to 50 mL tubes and centrifuged at 6000 g for 12 min. The supernatant was concentrated at 40 oC to a volume of
approximately 1 mL. The clean-up of the extract was carried out by solid-phase extraction on a 5 mL column (75×13 mm) packed successively with 2 g silica gel (0.063 – 0.200 mm), 1.5 g florisil and 0.65 g of anhydrous sodium sulfate. The packed column was fitted on an SPE vacuum manifold from Alltech (Deerfield, IL, USA). The column was conditioned with 6 mL dichloromethane and not allowed to dry. The extract (1 mL) was applied to the column and
the analytes were eluted with 45 mL n
-hexane-dichloromethane (3:2 v/v). The eluate was concentrated almost to dryness under a gentle stream of nitrogen and diluted with n-hexane to a volume of 1.0 mL. A 1 µL volume was injected automatically into the GC-MS.
Standard solutions
A standard stock mixture containing nine OCP was prepared in n-hexane at a concentration of 400 µg mL-1
and stored at -18 °C until use. The solutions were stable for at least two months, although it was verified that 4,4’-DDT and endrin are not stable in solution for three months and their degradation products were observed in chromatograms obtained in the full scan mode. The working standard solutions were prepared daily at concentrations of 0.160 to 21.0 µg mL-1 by dilution of the
standard stock solution with hexane.
Analytical curves
Determinations of the OCP were carried out by the external standard method using analytical curves with six
concentration levels in five replicates over the range of 0.160 to 21.0 µg mL-1.
Recovery studies
A volume of 1.0 mL of the spiking standard solutions containing the OCP mixture in n-hexane was added to 2.0 g dried and homogenized plant samples to obtain the fortification levels of 20, 40 and 200 ng g-1. The samples
were stored at room temperature one week before analysis for incorporation of the pesticides in the matrix and evaporation of the solvent. A minimum of two fortification levels with three replicates were prepared. All samples were treated and analyzed using GC-MS in the SIM mode, as previous described.
Method validation
The method was in-house validated through the
following performance criteria: linearity, linear range, sensitivity, limit of detection (LOD), limit of quantitation (LOQ), intra and inter-assay precision and accuracy.
Results and Discussion
The separation of hexachlorobenzene, lindane, heptachlor, aldrin, heptachlor epoxide, dieldrin, 4,4’-DDE, endrin and 4,4’-DDT was optimized for the GC-MS operation in the SIM mode, as previously described. Figure 2 shows the total ion chromatogram, after optimization of the chromatographic parameters, for a standard mixture containing the pesticides under study.
After establishment of the chromatographic conditions the method was in-house validated for OCP determination in plants. The linear range, linearity and sensitivity were established through analytical curves obtained by quintuplicate analyses of hexachlorobenzene, lindane, heptachlor, aldrin, heptachlor epoxide, dieldrin, 4,4’-DDE, endrin and 4,4’-DDT at six concentration levels in the range of 0.160 to 21.0 µg mL-1. The linearity, expressed
as the linear regression coefficients, were greater than 0.999 for all compounds under study, which is evidence of acceptable fits of the data to the regression line (Table 2). The sensitivity of the method for each compound was expressed as the slope of the analytical curve.
The sample preparation (extraction and clean-up) was based on the procedure recommended by Manirakiza et al.9 for the determination of organochlorine pesticides in
condiments by GC-ECD using, for clean-up, solid phase extraction with a mixed florisil/silica stationary phase and 10 mL n-hexane-dichloromethane (4:1 v/v) as elution solvent. Fortified samples of the three medicinal plants with the nine OCP under study were employed. Application of the conditions recommended by these authors resulted in a recovery lower than 50%. Consequently, the procedure was optimized in relation to the solvent strength and volume for the elution of the pesticides from the mixed SPE column. With 45 mL
n-hexane-dichloromethane (3:2 v/v), the recovery of all analytes, for a 2.0 g sample, was in the range of 70 to 120%.
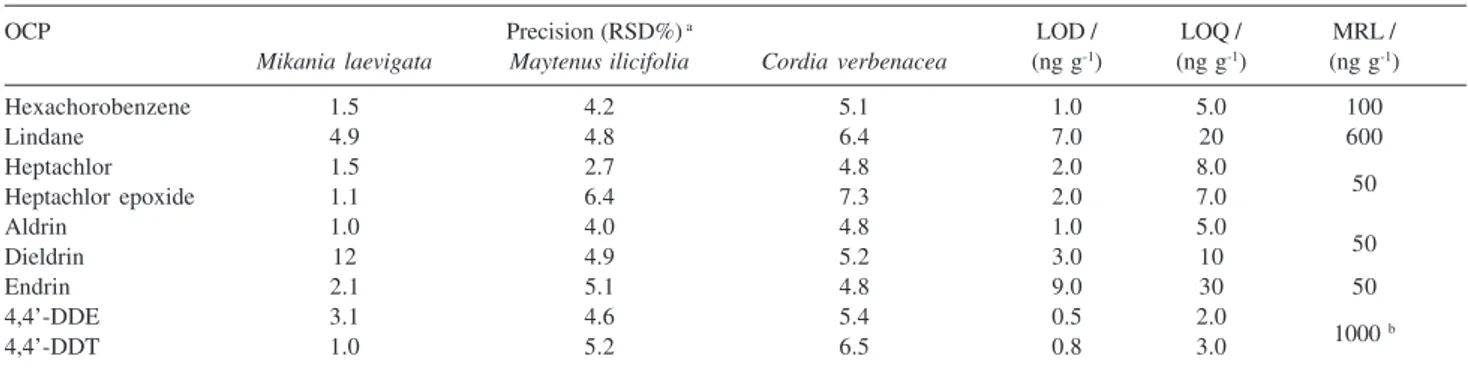
The inter-assay precision of the method was evaluated by the determination of the OCP, on six different days, in 2.0 g medicinal plant samples fortified with 0.4 µg OCP. The RSD ranged from 1.0 to 12%, as shown in Table 3,
which is in accordance with the value established by the Commission of the European Community21 or repeated
analysis of a sample carried out under within-laboratory reproducibility conditions, where the coefficient of variation shall not exceed 20% at this concentration level. The LOD and LOQ were determined at a signal-to-noise ratio of 3 and 10, respectively, measured at the approximate retention time of the corresponding analyte peak. LOD and LOQ ranged from 0.5 to 9.0 ng g-1 and 3.0
to 30 ng g-1, respectively (Table 3). All these values were
confirmed by the analyses of fortified samples of the plant samples at the respective OCP level. The method presents adequate detectivity and permits the determination of the pesticides with accuracy at levels lower than the MRL established in the European Pharmacopoeia for residues of OCP in medicinal plants (Table 3).
Due to the fact that no certified reference material is available, the accuracy of the method was evaluated through recovery tests, using fortified blank samples of
Mikania laevigata, Maytenus ilicifolia and Cordia
Figure 2. Total ion chromatogram obtained by GC-MS of OCP standards. (1) Hexachlorobenzene 5.11 µg mL-1; (2) Lindane 4.51 µg mL-1; (3) Heptachlor
4.53 µg mL-1; (4) Aldrin 4.47 µg mL-1; (5) Heptachlor epoxide 4.50 µg mL-1; (6) Dieldrin 4.42 µg mL-1; (7) 4,4’-DDE 4.05 µg mL-1; (8) Endrin
5.78 µg mL-1 and (9) 4,4’-DDT 4.54 µg mL-1.
Table 2. Linear range and linearity for the GC-MS determination
OCP Linear range/(µg mL-1) Linearity (r) Sensitivity/(au mL µg-1) RSD/(%)
Hexachlorobenzene 0.200 - 15.0 0.9998 17.0×103 3.00
Lindane 0.170 - 16.8 0.9999 6.97×103 3.60
Heptachlor 0.210 - 21.0 0.9998 6.38×103 2.07
Aldrin 0.160 - 16.2 0.9996 6.65×103 3.38
Heptachlor epoxide 0.160 - 16.2 0.9997 7.10×103 1.95
Dieldrin 0.170 - 17.1 0.9997 3.17×103 3.77
4,4‘ – DDE 0.190 - 18.9 0.9999 11.0×103 1.95
Endrin 0.170 - 17.4 0.9999 6.62×103 3.96
4,4‘ – DDT 0.190 - 18.7 0.9990 25.0×103 1.15
140 GC-MS Determination of Organochlorine Pesticides J. Braz. Chem. Soc.
verbenacea, at three concentration levels (20; 40 and 200 ng g-1). The first concentration level tested was below the
MRL of all pesticides under study. The recovery values determined were in the range of 70 to 125% and could be considered acceptable as functions of the analyte concentrations (Table 4).
Since quantitation of the pesticides was carried out using a confirmatory method, no additional study about the selectivity of the method was necessary.
The validated method was applied for pesticide residue determinations on three different crop species of Mikania
laevigata, Maytenus ilicifolia and Cordia verbenacea
collected from the experimental area of the CPQBA in Paulinia, SP, and two samples of Mikania sp purchased from a local retail market.
Dieldrin was detected in most samples (Table 5). A typical total-ion chromatogram obtained from Maytenus ilicifolia is shown in Figure 3. The confirmation of the identity of this pesticide in the samples was done by the ratios of the two qualifier ions m/z 237 and m/z 345, while
m/z 263 was employed for the quantitation in the single ion monitoring mode (Table 1).
Table 4. Recovery of OCP in fortified samples of Mikania laevigata, Maytenus ilicifolia and Cordia verbenacea
OCP Fortification Average Recovery / (%)a
Level / (ng g-1) Mikania laevigata Maytenu silicifolia Cordia verbenacea
Hexachlorobenzene 20 108 (1.5) 74 (14) 70 (9.3)
40 —- 97 (1.0)
—-200 70 (6.0) 113 (4.2) 93 (5.1)
Lindane 20 94 (4.9) 79 (7.0) 88 (10)
40 —- 99 (11)
—-200 70 (4.7) 110 (4.8) 83 (6.4)
Heptachlor 20 118 (1.5) 87 (8.3) 104 (12)
40 —- 105 (2.4)
—-200 77 (6.4) 105 (2.7) 77 (4.8)
Aldrin 20 89 (1.0) 86 (15) 71 (11)
40 —- 106 (2.9)
—-200 74 (5.8) 115 (4.0) 70 (4.8)
Heptachlor epoxide 20 107 (1.1) 99 (9.5) 111 (8.8)
40 —- 96 (3.2)
—-200 73 (5.5) 125 (6.4) 75 (7.3)
Dieldrin 20 71 (12) 89 (13) 85 (9.9)
40 —- 103 (5.7)
—-200 70 (12) 96 (4.9) 97 (5.2)
4,4’-DDE 20 107 (3.1) 88 (8.4) 79 (9.2)
40 —- 91 (3.5)
—-200 84 (7.8) 99 (4.6) 92 (5.4)
Endrin 20 120 (2.1) 89 (9.2) 100 (7.5)
40 —- 95 (2.8)
—-200 90 (5.6) 106 (5.1) 97 (4.8)
4,4’-DDT 20 118 (1.0) 78 (7.7) 74 (9.0)
40 —- 124 (5.2)
—-200 108 (9.0) 101 (5.2) 90 (6.5)
a In brackets the relative standard deviation of the determinations (n=3).
Table 3. Precision and limits of detection (LOD) and quantitation (LOQ) for the GC-MS method, as well as MRL according to the European
Pharmaco-poeia22
OCP Precision (RSD%) a LOD / LOQ / MRL /
Mikania laevigata Maytenus ilicifolia Cordia verbenacea (ng g-1) (ng g-1) (ng g-1)
Hexachorobenzene 1.5 4.2 5.1 1.0 5.0 100
Lindane 4.9 4.8 6.4 7.0 20 600
Heptachlor 1.5 2.7 4.8 2.0 8.0
50
Heptachlor epoxide 1.1 6.4 7.3 2.0 7.0
Aldrin 1.0 4.0 4.8 1.0 5.0
50
Dieldrin 12 4.9 5.2 3.0 10
Endrin 2.1 5.1 4.8 9.0 30 50
4,4’-DDE 3.1 4.6 5.4 0.5 2.0
1000 b
4,4’-DDT 1.0 5.2 6.5 0.8 3.0
Even though hexachlorobenzene was detected in all samples and different species analyzed, its concentration was below the LOQ of the method (5.0 ng g-1), as well as
below the MRL established by the European Pharmacopoeia (100 ng g-1). Furthermore, 4,4’–DDE was
detected in Mikania laevigata and Cordia verbenacea
from a region near Paulinia, at concentrations lower than 2.0 ng g-1 and in one sample of Mikania sp, purchased
from a local market.
Dieldrin was present in all the three different crop cultures from Paulinia at residue levels higher than the MRL established by the European Pharmacopoeia22 (50
ng g-1). One sample from the retail market was also
contaminated with dieldrin, however at a lower level than the MRL. Due to the fact that dieldrin is the major metabolite of aldrin, the MRL is established using the sum of both compounds.
Even though the use of organochlorine pesticides was banned in Brazil in 1985 and is no longer used in the cultivation area of the plants under this study, the contamination by dieldrin of all three plant species analyzed could be a consequence of the boundary transport from neighborhood areas, where these illegal pesticides are still in use.
The environmental pollution of Paulínia and its regions with heavy metals, fluorides, as well as organochlorine pesticides is well known and several reports emitted by CETESB (Companhia de Tecnologia de Saneamento Ambiental) are available.23,24 Considering that a significant
proportion of the organochlorine pesticides or their degradation products may remain in the soil as a persistent residue bound to the soil colloids, it is also possible that the presence of dieldrin in the plant species analyzed is due to past contamination of the soil and water of the area where these plants were cultivated.
Table 5. Organochlorine pesticide (OCP) levels in medicinal plants
Pesticides Concentration in dried weight , n = 3 / (ng g-1)
Paulínia Market 1 Market 2
Mikaniala Maytenus Cordia Mikania sp Mikania sp
evigata ilicifolia verbenacea
Hexachlorobenzene < 5.0 < 5.0 < 5.0 < 5.0 < 5.0
Lindane nd a nd a nd a nd a nd a
Heptachlor nd b nd b nd b nd b nd b
Aldrin nd c nd c nd c nd c nd c
Heptachlor epoxide nd d nd d nd d nd d nd d
Dieldrin 110 72 100 22 nd e
4,4’ - DDE < 2.0 nd e < 2.0 2.0 nd f
Endrin nd f nd f nd f nd g nd g
4,4’ - DDT nd g nd g nd g nd h nd h
nd = not detected; a < 7.0 ng g-1; b < 2.0 ng g-1; c < 1.0 ng g-1; d < 2.0 ng g-1; e < 0.5 ng g-1; f < 9.0 ng g-1; g < 0.8 ng g-1.
Figure 3. Total ion chromatogram of the analysis of Maytenus ilicifolia cultivated in Paulínia, showing in the insert, the selected peak of dieldrin used for
142 GC-MS Determination of Organochlorine Pesticides J. Braz. Chem. Soc.
Conclusions
The method developed is simple regarding to the extraction and clean-up of the extract and the use of a GC-MS system showed adequate selectivity and detectivity for the determination of OCP in medicinal plants. Limits of quantitation (LOQ) for the OCP were in the range of 2.0 to 30 ng g-1 (for 4,4’-DDE and endrin,
respectively), and achieved the maximum residue levels established by the European Pharmacopoeia.
Taking into consideration the increase in the use of medicinal plants, as well as the high residue levels of dieldrin found in the analyzed samples, it is recommended that a comprehensive study be conducted in order to evaluate the presence of OCP in medicinal plants commercialized in the Brazilian retail market, and to contribute to the protection of the consumers’ health.
Acknowledgments
The authors wish to acknowledge FAPESP (01/10887-5) and FINEP/RECOPE from Brazil for the financial support for this study and thank Professor C.H. Collins for language assistance.
References
1. Huie, C.; Anal. Bioanal. Chem. 2002, 373, 23.
2. Farias, M.R. In Farmacognosia: da Planta ao Medicamento; Simões, C.M.O.; Schenkel, E.P.; Gosnmann, G.; Mello, J.C.P.; Mentz, L.A.; Petrovick, P.R., eds., Editora da Universidade/ UFRGS: Porto Alegre, 2001, ch. 12.
3. Zuin, V.G.; Vilegas, J.H.Y.; Phytother. Res. 2000, 14, 73. 4. Aguilar, C.; Penalver, A.; Pocurull, E.; Ferre, J.; Borrull, F.;
Marcé, R.M.; J Chromatogr. A 1999, 844, 425.
5. Azevedo, D.D.; Lacorte, S.; Vinhas, T.; Viana, P.; Barcelo, D.;
J Chromatogr. A 2000, 879, 13.
6. Barriada-Pereira, M.; Concha-Grana, E.; Gonzalez-Castro, M.J.; Muniategui-Lorenzo, S.; Lopez-Mahia, P.; Prada-Rodríguez, D.; Fernandez-Fernandez, E.; J. Chromatogr. A
2003, 1008,115.
7. Cai, L.; Xing, J.; Dong, L.; Wu, C.; J. Chromatogr. A 2003, 1015, 11.
8. World Health Organization; Quality Control Methods for Medicinal Plant Materials, WHO: Geneva, 1998.
9. Manirakiza. P.; Covaci, A.; Schepens, P.; Chromatographia
2000, 52, 787.
10. Di Muccio, A.; Generali, T.; Barbini, D.A.; Pelosi. P.; Ausili, A.; Vergori, F.; Girolimetti, S.; J. Chromatogr. A 1997, 765,
61.
11. Holstege, D.M.; Scharberg, D.L.; Tor, E.R.; Hart, L.C.; Galey, F.D.A.; J. Assoc. Off. Anal. Chem. Int. 1994, 77,1263. 12. Vidal, J.L.M.; Arrebola, F.J.; Mateu-Sánchez, M.; J.
Chromatogr. A 2002, 959, 203.
13. Sloboník, J.; Ramalho, S.; Van Baar, B.L.M.; Louter, A.J.H.; Brinkman, U.A.T.; Chemosphere 2000, 41, 1469.
14. Miyahara, M.; Okada, Y.; Takeda, H.; Aoki, G.; Kobayashi, A.; Saito, Y.; J. Agric. Food Chem. 1994, 42, 2795.
15. Lacorte, S.; Guiffard, I.; Fraisse, D.; Barceló, D.; Anal. Chem.
2000, 72, 1430.
16. Lino, C.M.; Silveira, M.I.N.; J. Chromatogr. A 1997, 769, 275. 17. Camel, V.; Analusis 1998, 26, M99.
18. Wu, H.H.; Swoo, J.H.; Anal. Chim. Acta 2001, 428, 111. 19. Agência Nacional de Vigilância Sanitária. Resolução RDC no
17, 24/02, p.1-7. Brasília, 2000. http://www.anvisa.gov.br/legis/ resol/2000/17_00rdc.htm, accessed in August 2006. 20. Zuin. V.G.; Yariwake, J.H.; Bicchi, C.; J. Chromatogr. A 2003,
985, 159.
21. European Community, 2002, Commission Directive L 201/93. 2002/657/EC, Off. J. Eur. Community, L221/8.
22. European Pharmacopoeia, Conseil de l’Europe, Strasbourg, 2005.
23. Pompéia, S.L.; Martins, S.E.; Pereira, A.; Diagnóstico da
Contaminação e Plano de Recuperação de uma Mata Ciliar Degradada por Efluentes Industriais de Paulínia-SP, São Paulo, CETESB, 1993, p. 59.
24. http://www.greenpeace.org.br/toxicos/pdf/relatiorio shell gp1.doc, accessed in November 2003.
Received: February 23, 2006
Web Release Date: January 19, 2007