Atypical presentations of
neuromyelitis optica
Douglas Sato1,2, Kazuo Fujihara3
ABSTRACT
Neuromyelitis optica (NMO) is an inflammatory disease of central nervous system classically characterized by acute, severe episodes of optic neuritis and longitudinally extensive transverse myelitis, usually with a relapsing course. The identification of an autoantibody exclusively detected in NMO patients against aquaporin-4 (AQP-4) has allowed identification of cases beyond the classical phenotype. Brain lesions, once thought as infrequent, can be observed in NMO patients, but lesions have different characteristics from the ones seen in multiple sclerosis. Additionally, some AQP-4 antibody positive patients may present with a variety of symptoms not being restricted to optic neuritis and acute myelitis during the first attack or in a relapse. Examples are not limited to, but may include patients only with brain and/or brainstem lesions, narcolepsy with hypothalamic lesions or patients with intractable hiccups, nausea and vomiting. The prompt identification of NMO patients with atypical presentations may benefit these patients with institution of early treatment to reduce disability and prevent further attacks.
Key words: neuromyelitis optica, aquaporin 4, myelitis, optic neuritis, diagnosis, differential, nausea, vomiting, hiccup, brain diseases.
Apresentações atípicas da neuromielite óptica
RESUMO
Neuromielite óptica (NMO) é uma doença inflamatória do sistema nervoso central caracterizada classicamente por neurite óptica grave e mielite transversa longitudinalmente extensa, com um curso usualmente recorrente. A identificação do anticorpo detectado exclusivamente nos pacientes com NMO contra a aquaporina-4 (AQP-4) permitiu a identificação de casos além do fenótipo clássico. Lesões cerebrais, que antes eram descritas como infrequentes, podem ser observadas em pacientes com NMO, mas as lesões possuem características diferentes das lesões observadas na esclerose múltipla. Além disso, alguns pacientes positivos para o anticorpo contra a AQP-4 podem apresentar uma variedade de sintomas não restritos à neurite óptica e mielite aguda, seja durante o primeiro ataque seja em uma recorrência. Exemplos não estão limitados aos descritos a seguir, mas incluem pacientes com lesões cerebrais e/ou tronco cerebral, narcolepsia com lesões hipotalâmicas ou pacientes com quadros intratáveis de soluços, náusea e vômitos. A identificação rápida dos pacientes com NMO com apresentações atípicas pode beneficiar estes pacientes com a instituição precoce do tratamento a fim de reduzir a incapacidade e prevenir ataques subsequentes.
Palavras-Chave: neuromielite óptica, aquaporina 4, mielite, neurite óptica, diagnóstico diferencial, náusea, vômito, soluço, encefalopatias.
Correspondence
Douglas K. Sato Department of Neurology Tohoku University Graduate School of Medicine
1-1, Seiryomachi, Aobaku Sendai, Miyagi, 980-8574, Japan
E-mail: [email protected]
Received 18 August 2011 Accepted 26 August 2011
1Department of Neurology, Tohoku University Graduate School of Medicine, Sendai, Japan; 2Department of Neurology,
Faculty of Medicine - University of São Paulo, São Paulo SP, Brazil; 3Department of Multiple Sclerosis Therapeutics, Tohoku
University Graduate School of Medicine, Sendai, Japan.
Neuromyelitis optica (NMO) or De-vic’s disease is an inlammatory disease of central nervous system classically charac-terized by acute, severe episodes of optic
neuritis (ON) and longitudinally exten-sive transverse myelitis (TM)1. he article
pa-tient presenting with bilateral ON followed by TM and she deceased about a month after the monophasic opti-comyelitis2. Further studies with NMO cohorts revealed
that majority of NMO patients have a relapsing disease course1-4.
NMO has often been classiied as a demyelinating disease, which includes multiple sclerosis (MS). How-ever, unique clinical and laboratory features found in NMO patients like poor functional recovery and rare chronic progression, relatively common involvement of optic chiasm, spinal cord lesions longitudinally extending over three or more vertebral segments, absence of oligo-clonal immunoglobulin G (IgG) bands, and prominent elevation of glial ibrillary acidic protein (GFAP) levels in the cerebrospinal luids diferentiate NMO patients from other demyelinating diseases. Additionally, patholog-ical indings in NMO patients like perivascular deposi-tion of immunoglobulins and complement, severe astro-cytic damage (demonstrated by loss of AQP-4 and GFAP stainings) and a relative preservation of myelin strongly suggest that NMO is a clinical entity distinct from MS5,6.
he identiication of an autoantibody exclusively de-tected in NMO patients against aquaporin-4 (AQP-4)7,
a water channel richly expressed in the end-feet of astro-cytes8 (AQP-4 antibody, also known as NMO-IgG)
pro-vided an opportunity for a signiicant progress on un-derstanding the molecular mechanisms behind NMO pathology. Among others, the demonstration in animal models9,10 (induction of transfer experimental
autoim-mune encephalomyelitis accompanied by the administra-tion of immunoglobulins from AQP-4 antibody positive patients) that AQP-4 antibody activates complement, in-duces astrocytic damage and other features of NMO le-sions, with a relative preservation of myelin basic protein staining as previously reported in pathological studies with NMO patients has provided evidence in vivo that
AQP-4 antibody is pathogenic in the presence of T cell-mediated brain inlammation.
Since 2004, the availability for AQP-4 antibody assay at some laboratories has allowed identiication of cases beyond the classical phenotype with a diversity of cen-tral nervous system (CNS) lesions11. What was once
be-lieved as atypical may be part of a common phenotype after some accumulated experience under the availability of AQP-4 antibody testing and may prompt physicians to investigate the presence of AQP-4 antibody in these patients further beyond the classical NMO phenotype.
What is typical for NMO?
he deinition of typical NMO phenotype could be illustrated according to Wingerchuk’s revised diagnostic criteria (2006)12 as patients with ON, acute myelitis, and
at least two of the following three supportive criteria: [1] longitudinally extensive spinal cord lesions contig-uous over three or more vertebral segments; [2] lack of brain lesions in the magnetic resonance imaging (MRI) fulilling MS criteria at disease onset and [3] serum pos-itivity for AQP-4 antibody. From the accumulated pub-lications about NMO, it seems that the most common phenotype observed in the clinical practice has attacks of ON and TM, frequently associated with severe dis-ability relected clinically as severe visual deicit, parapa-resis or tetrapaparapa-resis with sensory level and sphincter dis-turbances. he clinical and laboratory indings in NMO are summarized in the Table.
As compared with Wingerchuk’s original diagnostic criteria of NMO (1999)1, the revised diagnostic criteria12
has incorporated three major changes: it included AQP-4 antibody as a supportive criterion, allowed brain lesions atypical for MS at disease onset and excluded the restric-tion of lesions to the optic nerve and spinal cord (about 15% of NMO patients had extra-optic-spinal CNS symp-toms). As the revised diagnostic criteria is stringent in order to provide a high speciicity, the concept of NMO spectrum disorders was also proposed, which included limited forms of NMO (monophasic or recurrent
lon-Table. Summary of clinical and laboratory indings in NMO patients.
Typical features Not typical, but commonly reported Uncommon, observed in few patients
Unilateral or bilateral ON with severe
visual impairment Brain lesions in the hypothalamus, corpus callosum, periventricular area and brain-stem. Lesions usually have distinct fea-tures of MS lesions
Hypersomnia, associated with bilateral hypothalamic lesions and low CSF hypo-cretin-1 levels
TM with longitudinally extensive
(3≥VS) and centrally located lesion Intractable hiccups, nausea and vom-iting, often lasting for over 48 hours Transient asymptomatic elevation of CK levels; observed few weeks prior to a NMO attack
AQP-4 antibody positivity in the serum Painful tonic spasm in the TM recovery period
gitudinally extensive myelitis with three or more verte-bral segment MRI spinal cord lesions, and recurrent or simultaneous bilateral ON), ON or myelitis associated with “typical” NMO brain lesions like hypothalamic, cal-losal, periventricular and brainstem lesions, ON or lon-gitudinally extensive myelitis associated with systemic auto-immune diseases and so-called Asian opticospinal MS12,13. However, the deinition of NMO spectrum
dis-orders did not cover patients with AQP-4 antibody pos-itivity without ON and longitudinally extensive myelitis attacks, like AQP-4 antibody seropositive patients pre-senting only with brain or brainstem symptoms.
What is not typical, but frequent in NMO
A number of lesion sites beyond optic nerve and spinal cord have been described in NMO. Below, we review each of these “atypical” features reported in NMO patients.
Brain lesions
he initial description of NMO was a patient only with ON and TM, with pathological demonstration of lesions also restricted to these two sites2, and the
occur-rence of brain lesions in NMO had been thought as in-frequent. his concept has been retained until recently, as the original criteria proposed in 1999 excluded pa-tients with symptoms implicating other CNS regions than optic nerve and spinal cord, and required brain MRI at onset being normal or not fulilling Paty criteria1. As
described earlier, the revised criteria considered patients with brain symptoms and those not meeting Paty criteria at disease onset, but ON and acute myelitis remained ab-solute in the NMO diagnostic criteria. hus, AQP-4 anti-body-positive cases with brain symptoms in the absence of ON and acute myelitis were beyond the scope of the revised criteria.
Since AQP-4 antibody assays have become widely available as a clinical test, the percentage of AQP-4 an-tibody-positive patients with brain lesions has ranged from 60 to 79% of patients14,15. Moreover, some of the
brain lesions seen in NMO are atypical for MS, such as those as longitudinally extensive brain lesions, extensive hemispheric lesions, resembling acute disseminated en-cephalomyelitis or posterior reversible encephalopathy syndrome, periventricular lesions and brainstem le-sions often contiguous with longitudinally extensive cer-vical cord lesions15. Another group has reported
mul-tiple patchy gadolinium enhancing lesions with blurred margin, described as “cloud-like enhancement” in the brain MRI of NMO patients16. It is also remarkable that
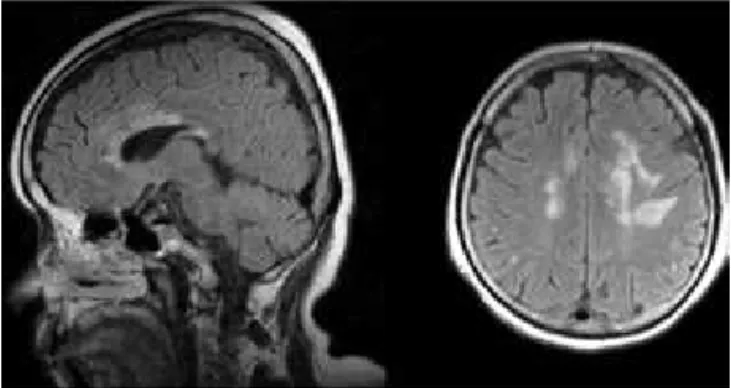
many of these lesion sites correspond with the regions with high expression of AQP-417. Fig 1 illustrates MRI
brain lesions in a NMO patient.
Callosal lesions
Although callosal lesions are part of brain lesions seen in NMO, they have characteristics that help difer-entiate them from the ones usually seen in MS. Callosal lesions detected in NMO patients with brain symptoms often have acute features such as contrast enhancement and difusion restriction14. hose acute lesions are
usu-ally large, multiple, and edematous with heterogeneous intensity described as a “marbled pattern”. Additionally, they can involve both anterior and posterior portions of the corpus callosum and extend into both cerebral hemispheres. Fig 2 shows a MRI from a NMO patient with acute callosal lesions. In the chronic stage, these cal-losal lesions observed in NMO patients frequently shrink or disappear. hese indings in NMO clearly difer from those of callosal lesions in MS, which are much smaller, often located at the callosal-septal interface in the middle and posterior thirds of the corpus callosum, and asym-metrical in the coronal sections.
Hiccups, nausea and vomiting
Intractable hiccup and nausea lasting over 48 hours seem to be a common feature in NMO. In our review of 144 cases of NMO spectrum disorders, 30 patients (21%) had hiccups and 24 (17%) had nausea18. In another study,
Fig 1. MRI Brain lesions in a patient with NMO. The initial symptom
of this attack was apathy and right lower limb paresis. Fig 2.
nearly a fifth of NMO patients presented with these symptoms whereas none of MS patients had reported such complains19. he episodes resolved spontaneously
or with methylprednisolone treatment. In most cases, brain MRI on sagittal planes detected linear medullary lesions located in the periaqueductal region including the area postrema, a putative vomiting center, and the nucleus tractus solitarius. hese medullary lesions, to-gether with long and centrally located myelitis are likely to be another characteristic feature of NMO spectrum disorders. Fig 3 demonstrates a MRI linear medullary lesion associated with intractable hiccups observed in a NMO patient.
Another study characterized the neuropathological features of NMO patients with intractable nausea and vomiting20. Forty percent of these patients exhibited
uni-lateral or biuni-lateral lesions involving the area postrema and the medullary loor of the fourth ventricle, and those lesions had pathological features previously reported in spinal cord pathology of NMO including loss of AQP-4 staining, preservation of myelin, and complement de-position. However, unlike spinal cord lesions in NMO, GFAP staining was retained in the medullary lesions, suggesting a milder involvement.
Painful tonic spasm
Painful tonic spasm is paroxysmal tonic muscle con-traction afecting one or more limbs that lasts less than a minute, and often occurs in severe myelopathy of NMO. he movement of the afected limbs usually triggers the painful tonic spasm. In our retrospective study of 37 tients with NMO and 68 patients with MS, 43% of pa-tients with NMO experienced painful tonic spasm while only 10% had the symptom in the MS group21. Painful
tonic spasm in NMO was more frequent and uncomfort-able than that in MS and required drug treatment more frequently. Painful tonic spasm is usually responsive to treatment with antiepileptic drugs like carbamazepine
administered in low doses, so the early diagnosis is im-portant for a proper management.
Uncommon clinical presentations in NMO
Hypersomnia – Excessive daytime sleepiness has been reported as a symptom of NMO spectrum disorder, as-sociated with low levels of hypocretin-1, a hypothalamic neuropeptide for sleep-wake regulation, in the cerebro-spinal luids as seen in narcolepsy22,23. Nocturnal
poly-somnography and multiple sleep latency test revealed that our patient’s sleep latency was less than ten min-utes with sleep onset REM periods and the total sleep time was over nine hours. he symptom is caused by bi-lateral hypothalamic lesions, and may resolve spontane-ously or shows a good response to corticosteroids treat-ment. his symptom can also be the irst episode / attack of NMO spectrum disorder.
HyperCKemia episodes – In our retrospective study with 736 AQP-4 antibody positive patients, three pa-tients (0.4%) with NMO had prominent elevation of CK levels (12.520, 19.415, and 59.660 IU/L) associated with general fatigue a few weeks before the onset of ON24.
Ele-vated CK levels resolved without any treatment in all pa-tients, but recurred once in one of them. Although skel-etal muscles express AQP-4 and this transitory elevation of CK levels may be immune-mediated, induce or stim-ulate AQP-4 autoimmunity, the direct relationship with AQP-4 antibody is still to be conirmed. Milder elevation on CK levels could be more common but unnoticed, and further studies are needed.
Final remarks – NMO patients may present a broader spectrum of symptoms than previously described in the classical descriptions. Further studies with large NMO cohorts may confirm the accumulated knowledge on case series studies and ind additional “atypical” features of NMO spectrum disorders. It is important to be aware that patients with NMO spectrum disorders may have an initial symptom outside of optic nerve and spinal cord, such as brain and brainstem attacks. Early treatment may reduce the number of further attacks and provide ben-eit to these patients.
ACKNOWLEDGMENTS – We would like to thank Dr Dagoberto Cal-legaro, University of São Paulo, for helpful comments on the manuscript.
REFERENCES
1. Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology 1999;53: 1107-1114.
2. Miyazawa I, Fujihara K, Itoyama Y. Eugène Devic (1858-1930). J Neurol 2002;249:351-352.
3. Collongues N, Marignier R, Zephir H, et al. Neuromyelitis optica in France: a multicenter study of 125 patients. Neurology 2010;74:736-742. 4. Adoni T, Lino AM, da Gama PD, et al. Recurrent neuromyelitis optica in
Bra-zilian patients: clinical, immunological, and neuroimaging characteristics. Mult Scler 2010;16:81-86.
5. Takano R, Misu T, Takahashi T, Sato S, Fujihara K, Itoyama Y. Astrocytic damage is far more severe than demyelination in NMO: a clinical CSF bio-marker study. Neurology 2010;75:208-216.
6. Misu T, Fujihara K, Kakita A, et al. Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain 2007;130: 1224-1234.
7. Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106-2112.
8. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005;202:473-477.
9. Kinoshita M, Nakatsuji Y, Kimura T, et al. Neuromyelitis optica: passive transfer to rats by human immunoglobulin. Biochem Biophys Res Commun 2009;386:623-627.
10. Bradl M, Misu T, Takahashi T, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol 2009;66:630-643. 11. Sato D, Fujihara K. Neuromyelitis optica without typical opticospinal
phenotype. Mult Scler 2010;16:1154-1155.
12. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006; 66:1485-1489.
13. Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007;6:805-815. 14. Pittock SJ, Lennon VA, Krecke K, Wingerchuk DM, Lucchinetti CF,
Wein-shenker BG. Brain abnormalities in neuromyelitis optica. Arch Neurol 2006;63:390-396.
15. Kim W, Park MS, Lee SH, et al. Characteristic brain magnetic resonance
imaging abnormalities in central nervous system aquaporin-4 autoimmu-nity. Mult Scler 2010;16:1229-1236.
16. Ito S, Mori M, Makino T, Hayakawa S, Kuwabara S. “Cloud-like enhance-ment” is a magnetic resonance imaging abnormality specific to neuro-myelitis optica. Ann Neurol 2009;66:425-428.
17. Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol 2006;63:964-968.
18. Sato D, Takahashi T, Nakashima I, Misu T, Itoyama Y, Fujihara K. Aqua-porin-4 antibody positive male patients: an analysis of 144 cases. Poster presented at the annual meeting of the American Academy of Neurology, Hawaii, HL 2011.
19. Misu T, Fujihara K, Nakashima I, Sato S, Itoyama Y. Intractable hiccup and nausea with periaqueductal lesions in neuromyelitis optica. Neurology 2005;65:1479-1482.
20. Popescu BF, Lennon VA, Parisi JE, et al. Neuromyelitis optica unique area postrema lesions: nausea, vomiting, and pathogenic implications. Neurology 2011;76:1229-1237.
21. Takai Y, Nakashima I, Misu T, Fujihara K, Itoyama Y. Painful tonic spasm in neuromyelitis optica. Neurology 2010;74:A168-A169.
22. Baba T, Nakashima I, Kanbayashi T, et al. Narcolepsy as an initial manifes-tation of neuromyelitis optica with anti-aquaporin-4 antibody. J Neurol 2009;256:287-288.
23. Kanbayashi T, Shimohata T, Nakashima I, et al. Symptomatic narcolepsy in patients with neuromyelitis optica and multiple sclerosis: new neurochem-ical and immunologneurochem-ical implications. Arch Neurol 2009;66: 1563-1566. 24. Suzuki N, Takahashi T, Aoki M, et al. Neuromyelitis optica preceded by